Part number 7. RNAInSpace - software for the semi-automatic construction of RNA in space
In Previous Parts From Proteins to RNA , Mat. Criteria , How to reduce the number of turns of the circuit? , How to evaluate the progress of folding single- RNA? , The limitation of optimizing methods in games with and without an opponent , One fundamental problem , Introduction to folding multi-helical RNAs, I told the basics of my proposed cybernetics-geometric approach for the task of folding RNAs . I repeat the formulation of the problem:
We have an arbitrary, really existing, primary sequence of up to 100 nucleotides. We know all the hydrogen bonds that need to be formed. The output is a .pdb file in which the tertiary structure is from the specified primary sequence and where all the required hydrogen bonds are formed.
Here I will talk about practice so that everyone can try what it is. I developed software to calculate what I was talking about. Here I give a link to the demo version . And I explain what you can see. It’s better to see once than to hear 100 times :)
After you download ( download from here), unzip it, and run RNAInSpace.exe, click on “Start” and you will see a screen (only with an empty top and bottom window) (an existing bug is known, sometimes the top window may not fall into place normally - just restart and repeat):
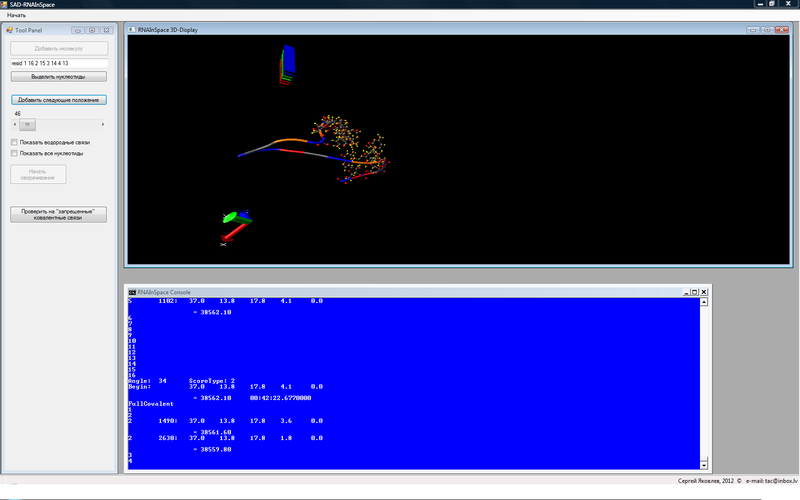
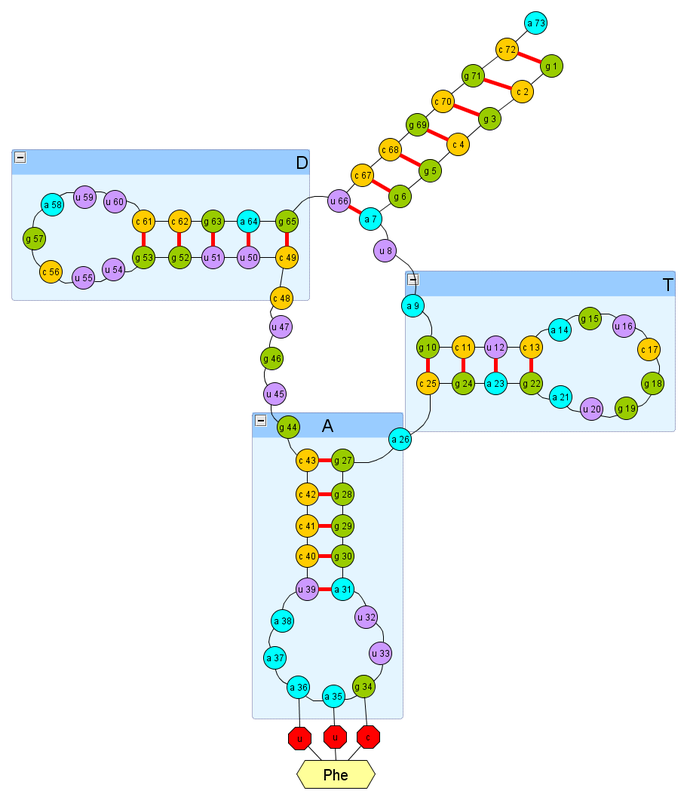
This is really a demo -version, you will not be able to control the folding algorithm. But otherwise, you can follow how folding is done. Initially, the version is configured to fold one helix of the T-loop of the transport RNA of the body of Escherichia coli, which transfers phenylalanine, below its secondary structure (the part labeled “T”):
Now how to manage the demo version.
The first thing you do after downloading is to click on the “Start Collapsing” button. Numbers will appear in the bottom screen of the console. About them later. Check in the directory where RNAInSpace.exe is located, the files X _ #. Pdb began to appear. As soon as X_0.pdb appeared, click on the “Add Molecule” button. On the screen at the top, you will see an RNA strand stretched in a line. You can click the "Select nucleotides" button. There will be 4 pairs of nucleotides that should form hydrogen bonds between themselves (but they can be changed by entering the nucleotide numbers above, after the word resid, which you want to display). The two CheckBox's below show / hide hydrogen bonds (if any) and all nucleotides of the chain.
As soon as the next X_1.pdb, X_2.pdb, X_3.pdb, etc. appears. you can click the "Add the following position" button and see how the RNA begins to fold slightly.
The graphic window can be expanded. As well as controlling the mouse, you can rotate the molecule (press the left / right button and move - rotates, turn the wheel to increase / decrease).
If you turn off the program and run it again, the folding does not start again, but from the last step at which the folding stopped the previous time. If you want to start from the very beginning, you need to clear the traj.dat file. And in order not to get confused, the next time you start, you should delete all the old X _ #. Pdb and X _ #. Txt files. You can also view the .pdb format in standard molecular imaging software, such as PyMOL .
The demo version does not guarantee that the spiral will curl up - it’s like luck :)
As soon as you play enough with the visual presentation and want to figure out what the numbers in the console mean (though if you carefully read past articles, you can guess) during the calculations and what else can be done on the demo program, or just ask questions, ask, in the next part I will try to summarize the questions and make the FAQ. Of course, if necessary, and there will be interest.
PS Maybe someone is not lucky, my laptop doesn’t load graphics until I figured out what the problem is. Write about your problems, as far as possible I will watch.
upd. I’m simultaneously testing this demo program. At step 192 (file X_192.pdb), an interesting situation just appears, which I described inPart number 5. One fundamental problem . There hydrogen bonds are formed between the ends of the helix (1-16 nucleotides) while the inside of the helix is not yet formed. Then they will tear and form first closer to the loop.
We have an arbitrary, really existing, primary sequence of up to 100 nucleotides. We know all the hydrogen bonds that need to be formed. The output is a .pdb file in which the tertiary structure is from the specified primary sequence and where all the required hydrogen bonds are formed.
Here I will talk about practice so that everyone can try what it is. I developed software to calculate what I was talking about. Here I give a link to the demo version . And I explain what you can see. It’s better to see once than to hear 100 times :)
After you download ( download from here), unzip it, and run RNAInSpace.exe, click on “Start” and you will see a screen (only with an empty top and bottom window) (an existing bug is known, sometimes the top window may not fall into place normally - just restart and repeat):
This is really a demo -version, you will not be able to control the folding algorithm. But otherwise, you can follow how folding is done. Initially, the version is configured to fold one helix of the T-loop of the transport RNA of the body of Escherichia coli, which transfers phenylalanine, below its secondary structure (the part labeled “T”):
Now how to manage the demo version.
The first thing you do after downloading is to click on the “Start Collapsing” button. Numbers will appear in the bottom screen of the console. About them later. Check in the directory where RNAInSpace.exe is located, the files X _ #. Pdb began to appear. As soon as X_0.pdb appeared, click on the “Add Molecule” button. On the screen at the top, you will see an RNA strand stretched in a line. You can click the "Select nucleotides" button. There will be 4 pairs of nucleotides that should form hydrogen bonds between themselves (but they can be changed by entering the nucleotide numbers above, after the word resid, which you want to display). The two CheckBox's below show / hide hydrogen bonds (if any) and all nucleotides of the chain.
As soon as the next X_1.pdb, X_2.pdb, X_3.pdb, etc. appears. you can click the "Add the following position" button and see how the RNA begins to fold slightly.
The graphic window can be expanded. As well as controlling the mouse, you can rotate the molecule (press the left / right button and move - rotates, turn the wheel to increase / decrease).
If you turn off the program and run it again, the folding does not start again, but from the last step at which the folding stopped the previous time. If you want to start from the very beginning, you need to clear the traj.dat file. And in order not to get confused, the next time you start, you should delete all the old X _ #. Pdb and X _ #. Txt files. You can also view the .pdb format in standard molecular imaging software, such as PyMOL .
The demo version does not guarantee that the spiral will curl up - it’s like luck :)
As soon as you play enough with the visual presentation and want to figure out what the numbers in the console mean (though if you carefully read past articles, you can guess) during the calculations and what else can be done on the demo program, or just ask questions, ask, in the next part I will try to summarize the questions and make the FAQ. Of course, if necessary, and there will be interest.
PS Maybe someone is not lucky, my laptop doesn’t load graphics until I figured out what the problem is. Write about your problems, as far as possible I will watch.
upd. I’m simultaneously testing this demo program. At step 192 (file X_192.pdb), an interesting situation just appears, which I described inPart number 5. One fundamental problem . There hydrogen bonds are formed between the ends of the helix (1-16 nucleotides) while the inside of the helix is not yet formed. Then they will tear and form first closer to the loop.