How we recreate our evolutionary past
- Transfer
An approach called " cladistics " works with living organisms, fossil remains and DNA.

How do we know what today's forms of life looked like when they first evolved? For years, biologists could draw conclusions about the common ancestors of today's life forms, on the basis of cladistics, which estimated the number of similar features. This approach worked with fossils and living beings, and allowed us to classify them into branching hierarchies that emanated from a common ancestor. But today we have DNA instead of bone shapes and the number of teeth.
How do we build a tree for her? It turns out that the general cladistic approach works here.
Suppose we need to understand the ancestors of mammals. For this you need to take a separate, but related group - reptiles are suitable for mammals. Reptiles and mammals have many similarities - four limbs (all of them are tetrapods, even snakes and whales with rudimentary limbs). There are differences - fur or special bones in the inner ear. There are partially similar features - laying eggs at the platypus, or features that are present only in a certain group - flying in bats.
Using these features, you can track how everything is grouped. Although bats and whales are very different, they do not lay eggs, therefore, most likely, they are more closely related to each other than with egg-laying. And since the latter lay eggs, like reptiles, they are likely to have branched off from the mammalian development tree rather early.
Sometimes strange features violate the slenderness of the tree - for example, both snakes and whales have only rudimentary limbs, despite the fact that for the rest of the signs they are weakly connected. Such problems are sometimes solved by collecting information about a sufficient number of features. The number of differences separating whales and other mammals is less than the number of similar features.
The logic of this analysis is simple: the more closely related the members of the group are, the fewer differences they have. Therefore, people clearly belong to primates, and not to rodents.
Kladistika can help sort out the past. Reptiles and ovipositories lay eggs, like duckbills, it can be assumed that at the time when the mammals separated the egg-laying mammals, all their ancestors also laid eggs. You can figure out what features first appeared in an already extinct mammal, and draw up an order of a branch from the development tree.
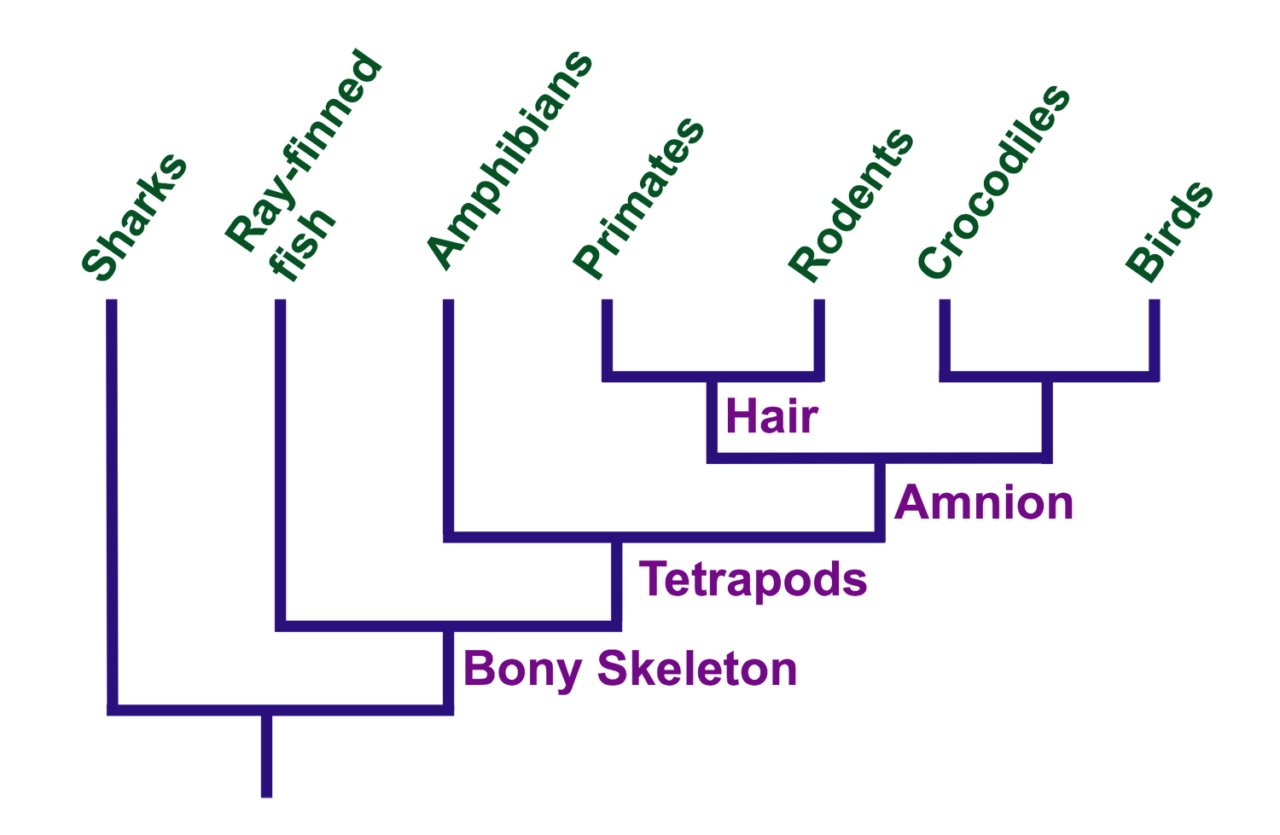
Sharks / Ray fish / Amphibians / Primates / Rodents / Crocodiles / Birds
Bottom up: bone skeleton, tetrapods, amnion, fur
It is important to note that such reasoning is not always perfect. For example, it is known that whales and hippos are fairly close relatives. They live in water, so it would be logical to assume that their ancestor lived there. However, the fossils say the opposite - they represent two different branches from ancestors who lived on land.
Conveniently, the same reasoning is suitable for DNA. Kladistika is based on the idea that organisms with minimal differences should be the closest related by kinship. Almost every paired base may change due to mutation (with the exception of the base of the most important genes). Therefore, each pair can become a distinctive feature. A small number of changes requires a small number of mutations, so it most likely means a small difference between DNA sequences.
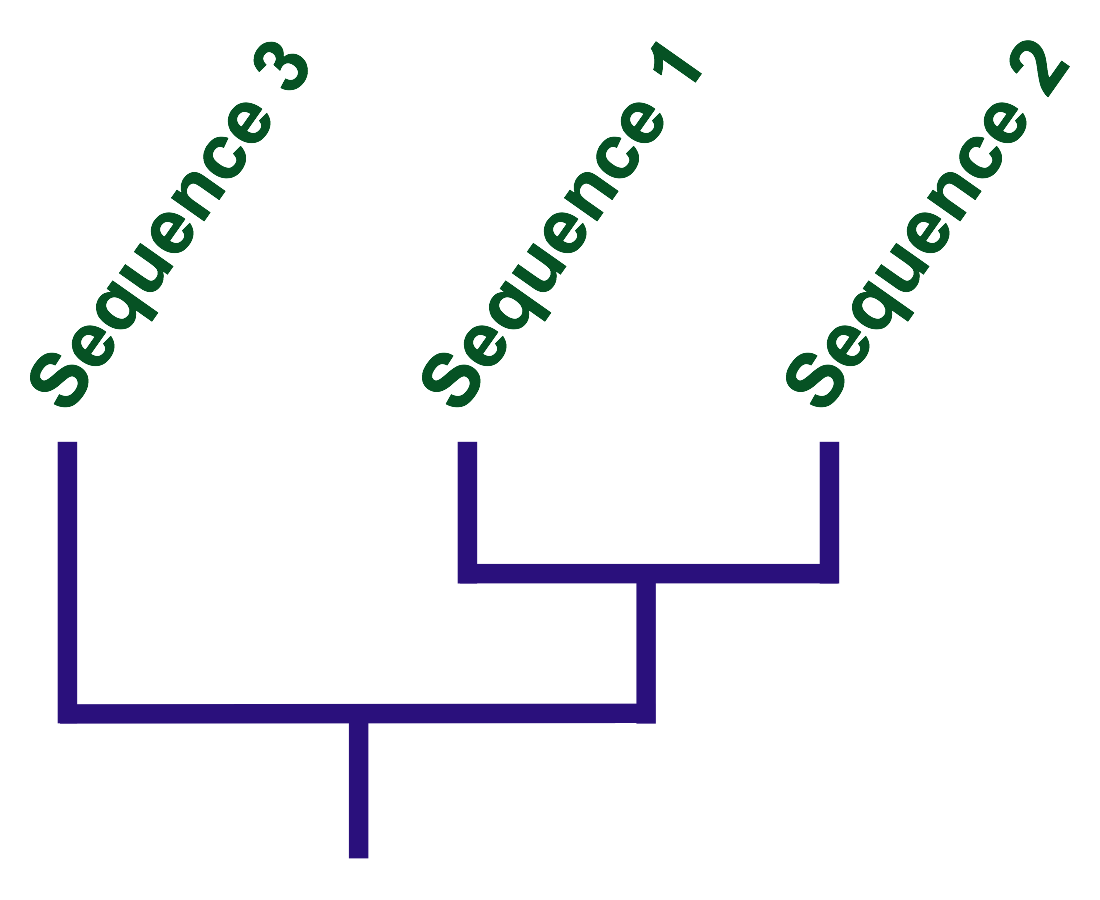
The three sequences in the picture below are different, and the difference in the second and third relative to the first is marked in red. There are only two differences between the first and second, and four between the first and the third. This means that the first two sequences are connected by closer ties.

And which of the first two sequences is closer to the third? To do this, repeat the process, just note the differences with respect to the third sequence.

It can be seen that the first and second have the same number of differences with the third. You can not say which of them is closer. A tree built on the basis of these relationships will look like this:
Researchers, finding out the relationship of two organisms, rely on something more than just a dozen of paired bases, but, in fact, the process is similar. You need to place a sequence on the intended tree and see how many changes are required for each of the branches. Shuffle the tree and repeat the analysis. As a result, the task is reduced to a simple search for a tree that requires the least amount of changes.
Well, or almost simple. One of the problems is the definition of equivalent sequences. Many vertebrate genes belong to larger genetic families. People have 22 different members of the FGF gene family. Comparing them with mice, it is necessary to compare human FGF-14 with the mouse equivalent of FGF-14, and not with other members of the family. Many of them can be lost or duplicated, and then the species can have two genes like FGF-14, or none.
These events — duplication and deletion of sequences — are constantly occurring outside the genes. When comparing different sequences, you need to understand that some of the reasons may not change, and the abyss. Other sequences may experience insertion of bases, which must also be taken into account. But with enough DNA and careful analysis, you can recreate evolutionary trees as well as with the features of the organisms.
However, you can recreate the past. Take a look at the same three sequences, marked up differently.

Now the red bases are marked, differing from the statistically most common bases of these three sequences. If we have evidence that these sequences had a common ancestor, then, most likely, these differences are single mutations in the past. If so, then we can assume the sequence of their ancestor - it requires the least amount of changes to obtain descendants. Consequently, if on the first basis two of the three sequences have G, and one of them has A, then their ancestor most likely had G.
And such an analysis has its limitations. It is impossible to know whether the foundation has changed in the entire history of existence more than once - and this becomes a problem when immersed in the distant past. But the technology works. We used it to recreate the ancient proteins, many of which are able to function in modern conditions. In many cases, they have different properties — say, the tolerance of high temperatures — that can tell us about the habitat of organisms.
It is also possible to use this to answer the question of when the ancestor protein existed or the species that used it. For most pedigrees, you can calculate the number of new mutations, the occurrence of which is typical for each generation. It can be compared with the total number of mutations and count the number of generations required to achieve the sequence of the current state. Adding the average time between generations, you can estimate the time required for the production of today's DNA. The errors are large, but this approach has its advantages.
“Not to return the past” is a cliche. But the past obviously leaves its mark on the present. Reading the tracks, you can make a vivid and informative picture of the past, even without a time machine to go there personally.
Note translate: An interesting example of the use of technology for estimating the time taken for the emergence of modern DNA is presented in the scientific work of Kittler, Kaiser and Stoneking . It is known that lice living in the hair on a person’s head, and lice living in his clothes are relatives, albeit distant. It turns out that it is possible to calculate how long their genetic paths diverged - and this date will characterize the moment when people began to put on clothes en masse. Scientists estimate that it happened about 72,000 years ago. True, the error is considerable: ± 42,000 years.
How do we know what today's forms of life looked like when they first evolved? For years, biologists could draw conclusions about the common ancestors of today's life forms, on the basis of cladistics, which estimated the number of similar features. This approach worked with fossils and living beings, and allowed us to classify them into branching hierarchies that emanated from a common ancestor. But today we have DNA instead of bone shapes and the number of teeth.
How do we build a tree for her? It turns out that the general cladistic approach works here.
Kladistika
Suppose we need to understand the ancestors of mammals. For this you need to take a separate, but related group - reptiles are suitable for mammals. Reptiles and mammals have many similarities - four limbs (all of them are tetrapods, even snakes and whales with rudimentary limbs). There are differences - fur or special bones in the inner ear. There are partially similar features - laying eggs at the platypus, or features that are present only in a certain group - flying in bats.
Using these features, you can track how everything is grouped. Although bats and whales are very different, they do not lay eggs, therefore, most likely, they are more closely related to each other than with egg-laying. And since the latter lay eggs, like reptiles, they are likely to have branched off from the mammalian development tree rather early.
Sometimes strange features violate the slenderness of the tree - for example, both snakes and whales have only rudimentary limbs, despite the fact that for the rest of the signs they are weakly connected. Such problems are sometimes solved by collecting information about a sufficient number of features. The number of differences separating whales and other mammals is less than the number of similar features.
The logic of this analysis is simple: the more closely related the members of the group are, the fewer differences they have. Therefore, people clearly belong to primates, and not to rodents.
Kladistika can help sort out the past. Reptiles and ovipositories lay eggs, like duckbills, it can be assumed that at the time when the mammals separated the egg-laying mammals, all their ancestors also laid eggs. You can figure out what features first appeared in an already extinct mammal, and draw up an order of a branch from the development tree.
Sharks / Ray fish / Amphibians / Primates / Rodents / Crocodiles / Birds
Bottom up: bone skeleton, tetrapods, amnion, fur
It is important to note that such reasoning is not always perfect. For example, it is known that whales and hippos are fairly close relatives. They live in water, so it would be logical to assume that their ancestor lived there. However, the fossils say the opposite - they represent two different branches from ancestors who lived on land.
Kladistika penetrates genetics
Conveniently, the same reasoning is suitable for DNA. Kladistika is based on the idea that organisms with minimal differences should be the closest related by kinship. Almost every paired base may change due to mutation (with the exception of the base of the most important genes). Therefore, each pair can become a distinctive feature. A small number of changes requires a small number of mutations, so it most likely means a small difference between DNA sequences.
The three sequences in the picture below are different, and the difference in the second and third relative to the first is marked in red. There are only two differences between the first and second, and four between the first and the third. This means that the first two sequences are connected by closer ties.
And which of the first two sequences is closer to the third? To do this, repeat the process, just note the differences with respect to the third sequence.
It can be seen that the first and second have the same number of differences with the third. You can not say which of them is closer. A tree built on the basis of these relationships will look like this:
Researchers, finding out the relationship of two organisms, rely on something more than just a dozen of paired bases, but, in fact, the process is similar. You need to place a sequence on the intended tree and see how many changes are required for each of the branches. Shuffle the tree and repeat the analysis. As a result, the task is reduced to a simple search for a tree that requires the least amount of changes.
Well, or almost simple. One of the problems is the definition of equivalent sequences. Many vertebrate genes belong to larger genetic families. People have 22 different members of the FGF gene family. Comparing them with mice, it is necessary to compare human FGF-14 with the mouse equivalent of FGF-14, and not with other members of the family. Many of them can be lost or duplicated, and then the species can have two genes like FGF-14, or none.
These events — duplication and deletion of sequences — are constantly occurring outside the genes. When comparing different sequences, you need to understand that some of the reasons may not change, and the abyss. Other sequences may experience insertion of bases, which must also be taken into account. But with enough DNA and careful analysis, you can recreate evolutionary trees as well as with the features of the organisms.
However, you can recreate the past. Take a look at the same three sequences, marked up differently.
Now the red bases are marked, differing from the statistically most common bases of these three sequences. If we have evidence that these sequences had a common ancestor, then, most likely, these differences are single mutations in the past. If so, then we can assume the sequence of their ancestor - it requires the least amount of changes to obtain descendants. Consequently, if on the first basis two of the three sequences have G, and one of them has A, then their ancestor most likely had G.
And such an analysis has its limitations. It is impossible to know whether the foundation has changed in the entire history of existence more than once - and this becomes a problem when immersed in the distant past. But the technology works. We used it to recreate the ancient proteins, many of which are able to function in modern conditions. In many cases, they have different properties — say, the tolerance of high temperatures — that can tell us about the habitat of organisms.
It is also possible to use this to answer the question of when the ancestor protein existed or the species that used it. For most pedigrees, you can calculate the number of new mutations, the occurrence of which is typical for each generation. It can be compared with the total number of mutations and count the number of generations required to achieve the sequence of the current state. Adding the average time between generations, you can estimate the time required for the production of today's DNA. The errors are large, but this approach has its advantages.
“Not to return the past” is a cliche. But the past obviously leaves its mark on the present. Reading the tracks, you can make a vivid and informative picture of the past, even without a time machine to go there personally.
Note translate: An interesting example of the use of technology for estimating the time taken for the emergence of modern DNA is presented in the scientific work of Kittler, Kaiser and Stoneking . It is known that lice living in the hair on a person’s head, and lice living in his clothes are relatives, albeit distant. It turns out that it is possible to calculate how long their genetic paths diverged - and this date will characterize the moment when people began to put on clothes en masse. Scientists estimate that it happened about 72,000 years ago. True, the error is considerable: ± 42,000 years.