Life and death of mitochondria
Mitochondria - small workers or big bosses?
If you think that the most important story of living together for us begins during a wedding, then this is not at all the case. The most important story of the joint life of each person began more than a billion years ago, when our distant unicellular ancestors were forced to sign a “marriage contract” with those whom we now call mitochondria (see the theory of symbiogenesis).
Mitochondria have two membranes (internal and external) and their own hereditary material in the form of DNA (Fig. 1). On the inner membrane of the mitochondria is a system of oxidative phosphorylation, which provides the oxidation of energy substrates with the formation of ATP.

Fig. 1 . The schematic structure of the mitochondria.
In the marriage contract of the cell and mitochondria there is no point “in sickness and health,” and that’s good. If the mitochondria becomes old, the cell can kill it in the process of mitophagy, and the mitochondria, in turn, regulate the process of apoptosis in incapable and old cells. If the process of mutual quality control is violated, the mechanisms of aging are triggered. The mechanisms of apoptosis are violated, the number of free radicals that are not controlled by the mitochondria increases. This causes systemic inflammation, cell DNA damage. Thus, there is a strong relationship between MX dysfunction, age-related diseases, aging of the body and metabolic dysfunctions [1]. Metabolic dysfunction - the constant rider of the apocalypse of aging.
Not all the blame for metabolic disorders lies with our overeating. Metabolic disorders are associated primarily with the inability of mitochondria to cope with nutrients. Mitochondria in a cell is not easy. We “feed” our cells either too much, then too little, and we present them with a “request” to give out energy in the form of ATP, the amount of which must exactly correspond to our needs. In order to “wriggle out” from this situation regularly, the mitochondria really use some “movements” - fission and fusion. These "mitodizations" unite under the name "dynamics of mitochondria". The balance between mitochondrial division and fusion is the central mechanism of bioenergetic adaptation to the metabolic needs of the cell [2, 3].
Most of all mitochondria are found in tissues with high energy needs - muscles, liver, brown adipose tissue, brain. It is not surprising that the dynamics of mitochondria in these tissues is better studied.
So, if a cell of any of these tissues (except for some neurons in the brain, about this later) receives a large amount of nutrients (intake exceeds costs), then the mitochondria are in a divided (fragmented) state. If the cell is in a state of hunger (revenues are less costly), then mitochondrial fusion occurs and they are in the United state. [3,4]. This is how cell homeostasis is maintained (Fig. 2).
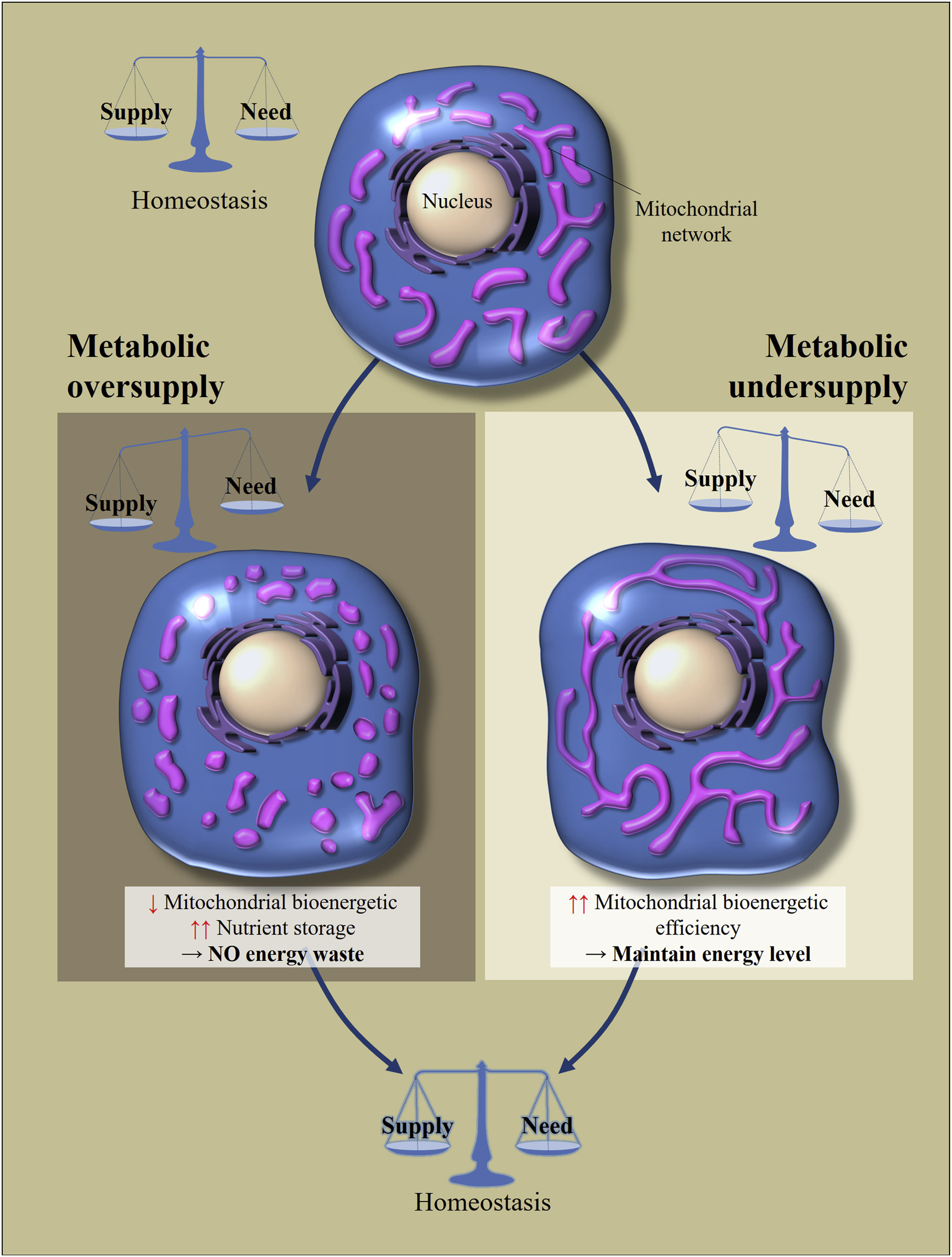
Fig. 2 Regulation of morphology and bioenergetic efficiency of mitochondria in response to excessive or insufficient nutrient intake [of 2]
Cellular metabolic homeostasis depends on the balance between the consumption of nutrients and their consumption. Changes in nutrient supply leads to cellular adaptations to restore balance. Excess nutrition leads to fragmentation of the mitochondrial network, which causes a decrease in the bioenergy efficiency of mitochondria. This will avoid energy loss. In contrast, with metabolic hunger, mitochondria are lengthened to increase their bioenergetic efficiency.
What is the trick of these movements? If the cell is in a state of starvation, the mitochondrial fusion allows increasing their bioenergetic efficiency (the amount of ATP that is created per nutrient molecule). If an excess of nutrients enters the cell, then they can either be 1) stored or 2) this energy can be dissipated as heat. The task of mitochondria in this case is to dissipate more energy in the form of heat, to store less in the form of ATP (accumulation of NADH and ROS will lead to oxidative stress). Fragmentation of mitochondria allows them to reduce bioenergetic efficiency, the main mechanism for reducing which is considered "leakage" of protons.
So, we go to work, and the life of the mitochondria constantly proceeds in the division and fusion cycle mode (Figure 3).
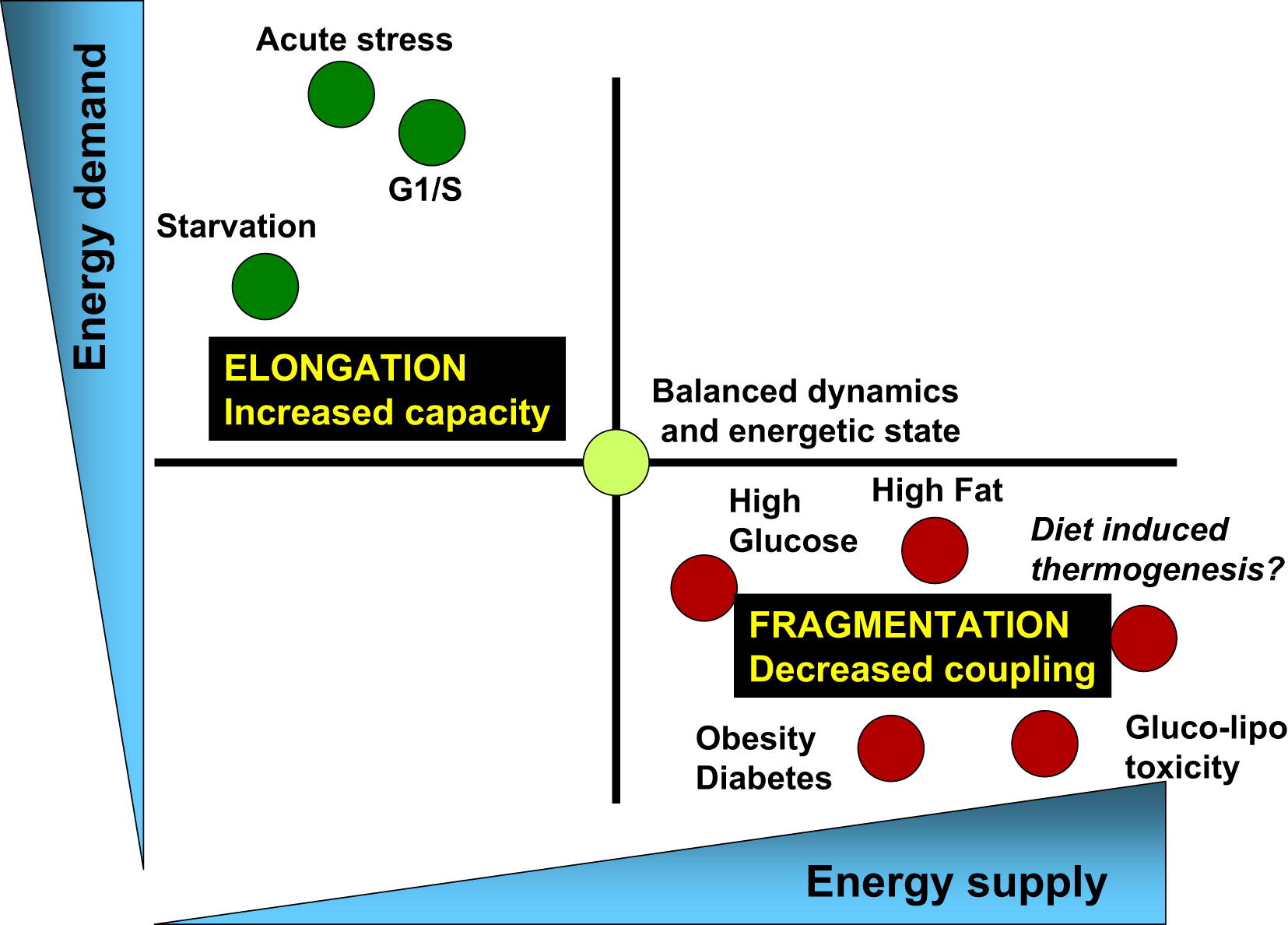
Pic.3The balance of energy consumption and energy is associated with the corresponding changes in mitochondrial architecture and their bioenergy efficiency [of 3]
Physiological processes associated with an increase in energy demand and reduced energy supplies (for example, acute stress, starvation and the G1 / S phase) are characterized by elongation of mitochondria and respiration associated with the synthesis of ATP. On the other hand, the physiological processes associated with a decrease in energy demand and an increase in its supply (high nutrient levels, obesity, and type 2 diabetes) are associated with mitochondrial fragmentation, heat release, or a decrease in mitochondrial function.
The normal cycle of mitochondrial division and their fusion is a key element in their quality control. Why? When mitochondrial division occurs, two daughter cells form, one of which has a higher membrane potential and goes further into the fusion-division cycle, while the other, with a more depolarized membrane, remains separated until the membrane potential is restored. If the potential is restored, it is reunited with the mitochondrial network. If it remains depolarized, then it is eliminated in the process of autophagy, which is the key to the quality of the mitochondrial pool (Fig. 4).
Long-term inhibition of mitochondrial division (with prolonged cell starvation) leads to the accumulation of damaged mitochondria, which cannot be segregated [3, 4].
On the other hand, an excess of nutrients leads to inhibition of mitochondrial fusion, which leads to disruption of the cycle of mitochondrial dynamics, increases intracellular mitochondrial heterogeneity. Yes, with an excess of food, mitochondrial fragmentation is protective, but long-term fragmentation, like long-term fusion, is detrimental to the quality control of mitochondria. There is no selective removal, the mitochondrial mass will decrease and consist of small depolarized mitochondria.

Fig.4. The life cycle of mitochondria and its regulation by the availability of nutrients [of 3]
At the molecular level, mitochondrial fusion is a two-step process that requires a coordinated fusion of the outer and inner membranes during individual successive events. In mammals, this process is regulated by three proteins that belong to GTPases: Mfn1 and Mfn2 are necessary for the fusion of the outer membrane, and OPA1 for the fusion of the inner membrane. Other proteins are needed for division, Fis1 and Drp1.
The role of mitofusin proteins has been studied in loss-and gain-of function studies. Mice mutant for mitofusin proteins die even in mid-gestation, because in them the fusion of mitochondria becomes impossible. Mitofuzins are important for autophagy and mitophagy. Reducing the expression of Mfn2 in cardiomyocytes blocks the start of the autophagy process, because the fusion of autophagosomes with lysosomes is blocked. The depletion of Mfn2 leads to a decrease in the potential of mitochondrial membranes, to compensate, the respiratory chain decreases, glucose uptake increases and glycogen synthesis decreases. The cell switches to anaerobic glyoclycine, and this is the path to oncological cell degeneration. A deficiency of Mfn2 leads to neurodegenerative changes. Increased expression of Mfn2 in skeletal muscle increases their insulin sensitivity.
Mfn1 performs similar functions, however, probably in other tissues (expression of Mfn2 and Mfn1 differs in different tissues) - Mfn1 is more expressed in the heart, liver, pancreas, testicles, and Mfn2 in the heart, skeletal muscles, brain, brown adipose tissue .
Thus mitofusins are key regulators of mitochondrial dynamics. The expression of mitofusins is different in different organs, they provide bioenergetic efficiency and adaptation mechanisms to the availability of nutrients, and the “fate” of the cell depends on them. Not surprisingly, mitochondrial fusion proteins are potential targets for pharmacological interventions [2, 5].
The dynamics of mitochondria is important in all cells. In the beta cells of the pancreas, mitochondria are nutrient sensors and signal generators of insulin synthesis, in muscles, the dynamics of mitochondria are important for the regulation of glucose metabolism, etc. However, a person is not just a collection of cells of a different type, each of which makes independent decisions. An organism is a system that has a central regulatory link for maintaining energy and glucose homeostasis. This major regulator is the hypothalamus.
The hypothalamus is located in the diencephalon and it is it that provides the interconnection of the nervous and humoral regulation systems. The hypothalamus neurons perceive, process and respond to signals from adipose tissue (leptin), pancreas (insulin), and other hormonal stimuli (ghrelin, cholecystokinin, pancreatic polypeptide, etc.). The hypothalamus controls the activity of the human endocrine system due to the fact that its neurons are able to secrete neuroendocrine transmitters that stimulate or suppress the production of hormones by the pituitary gland. In other words, the hypothalamus, whose mass does not exceed 5% of the brain, is the center of regulation of endocrine functions and maintaining homeostasis of the whole organism.
Even Dilman (Dilman V. M "Great biological clock") pointed to the leading role of the hypothalamus in the systematic development of metabolic dysfunction, leading to obesity, diabetes, cardiovascular diseases, cancer and aging. According to Dilman’s theory of hyper-adaptation, the sensitivity of the hypothalamic receptors to signals from the body’s tissues (leptin, insulin, etc.) gradually decreases gradually with age. In order to evoke his “answer” you need more and more of this or that hormone, more insulin, more leptin. Insulin and leptin resistance, metabolic diseases leading to aging and death are developing.
Depending on the functions performed, groups of neurons are combined into hypothalamic nuclei. One of them - arcuate (arcuate) core is a key regulator of eating behavior and metabolism. It can form orexigenic neuropeptides (stimulate appetite) and anorexigenic (suppress appetite), related, respectively, to AgRP and POMC neurons. Peripheral signals (insulin, ghrelin, leptin, etc.) affect the expression of peptides that stimulate or suppress the appetite, which ensures coherence of the central regulation (Fig. 5).

Fig. five.Hypothalamic control of energy metmbolism. The brain integrates metabolic signals (leptin, insulin, ghrelin, PYY3-36) from peripheral tissues, such as pancreas, adipose tissue, stomach. In the brain, specialized neural networks coordinate adaptive changes in the absorption and consumption of food [of 5].
The study of the dynamics of mitochondria in the brain tissues showed that the dynamics of mitochondria plays a significant role in the ability of the hypothalamus neurons to control the level of glucose and energy homeostasis in the body [6,7,8].
In AgRP neurons (hunger-promoting AgRP neurons), which stimulate appetite and regulate weight gain, starvation leads to the division of mitochondria, and high-fat feeding leads to fusion. That is, the mitochondrial response is different from that in most other cells.
The fusion of MX in these neurons regulates the electrical activity in response to a high-fat diet, stimulating the production of orexigenic peptide (AgRP peptide); it is necessary for weight gain and fat deposition with an excess of nutrients. Deletions of Mfn1 and Mfn2 in these neurons resulted in less weight gain in rats by reducing the level of circulating leptin.
POMC neurons (suppress appetite) have the opposite function, and the dynamics of mitochondria in response to the intake of nutrients they have different. Reduced expression of mitofusins in these neurons leads to disruption of the association of mitochondria with EPS, and, as a result, hyperphagia, leptin resistance and obesity. At the same time, food consumption increased, while energy consumption decreased.
Thus, the body's response to a high-fat diet depends on the patterns of mitochondrial dynamics in the hypothalamus neurons. Remodeling of mitochondria in neurons provides their response to the intake of nutrients into the body, stimulates the production of neuropeptides that will either stimulate or suppress appetite, affecting metabolism at the level of the organism (Fig.6).
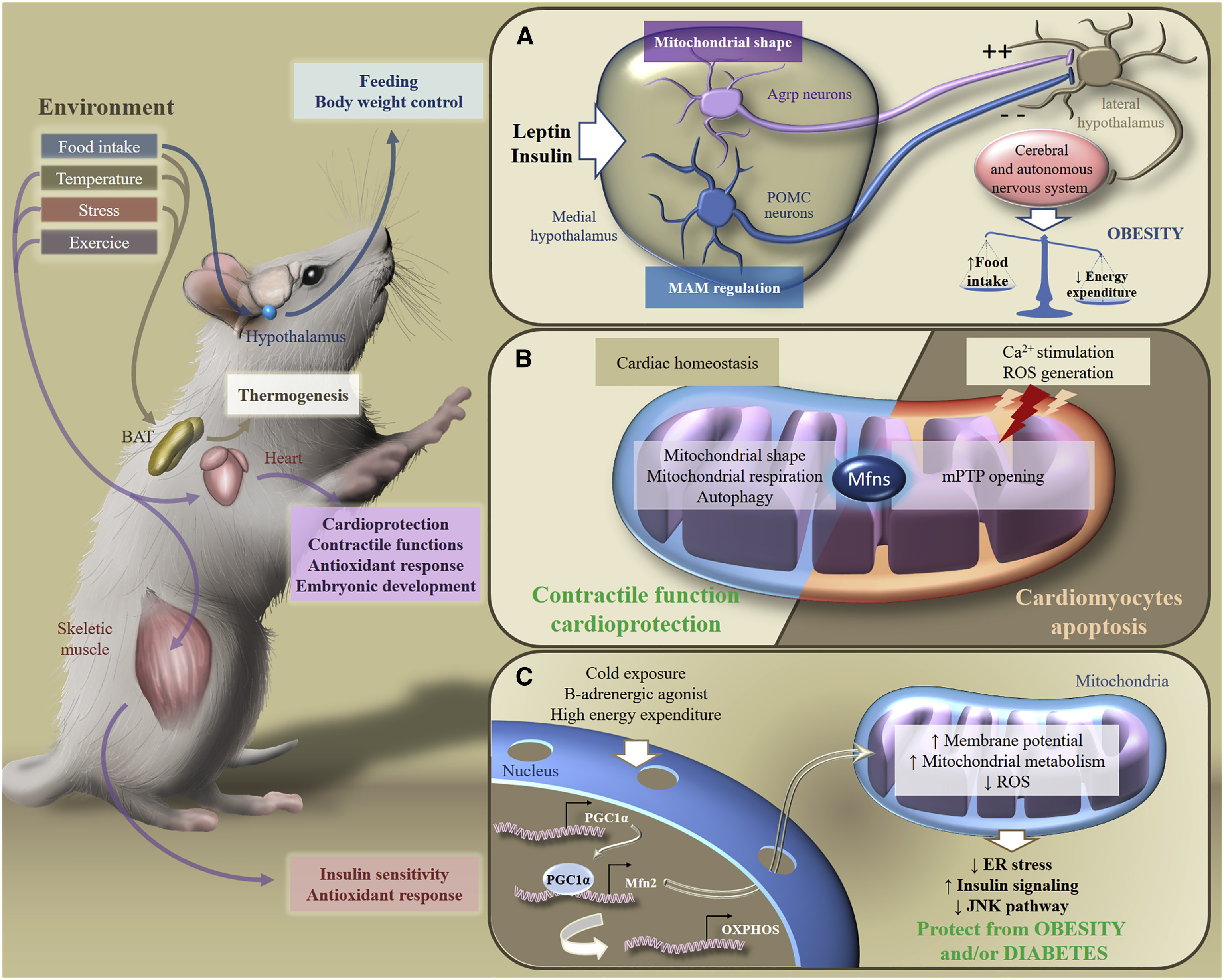
Fig.6. Metabolic adaptation to environmental stimuli [of 2]
In response to exogenous stimuli, Mfns are involved in the transduction of metabolic signaling in various organs, which ensures the maintenance of homeostasis of the whole body's energy. In particular, in response to food intake, temperature changes, stress or exercise, brown adipose tissue, the brain, heart or skeletal muscles adapt their metabolism to control nutrition, body weight, contractile functions, antioxidant response or insulin sensitivity.
1. Nutrition and Exercise
Nutrition Cycles Excess food and a high-fat diet (HFD) inhibit mitochondrial fusion in cells (in some neurons of the brain, the mechanism is different). An incomplete mitochondrial division-fusion cycle disrupts autophagy → the intracellular heterogeneity of mitochondria increases → no selective removal of mitochondria occurs → the mitochondria with dysfunction accumulate.
Calorie restriction (fed / fasting cycle) stimulates bioenergetic adaptation, ensuring the functioning of mitochondrial quality mechanisms.
2. Healthy membranes: stearic acid, cardiolipin, phosphatidic acid
All key processes depend on the “health” of mitochondrial membranes — autophagy, mitophagy, apoptosis, the association of mitochondria with the endoplasmic reticulum, and the dynamics of mitochondria. Cell membranes are composed of lipids and proteins. The remodeling of these membranes is controlled by the interactions between specific lipids and proteins.
Palmitic (C16) and stearic (C18) are among the saturated fatty acids. Stearic acid (C18: 0) has been shown to stimulate mitochondrial fusion. Its action is associated with the effect on mitofuzina. In mice, dietary supplements of stearic acid can partially restore mitochondrial dysfunction caused by mutations in the Pink1 or parkin genes. In the neutrophils of people who are on a low-C18: 0 diet for 2 days, the mitochondria are in a fragmented state (50% of the cells had fragmented MX, 10% united MX). Their use of stearic acid led to mitochondrial fusion after 3 hours [8]. Thus, steric acid is important for maintaining cycles of mitochondrial dynamics. Most of the stearic acid is in cocoa beans (31-34%).
Phospholipids are the main components of organelle membranes. They also regulate the dynamics of mitochondria, and their influence is different [9].
Cardiolipin (CL) stimulates mitochondrial division and fusion of internal membranes.
Cardiolipin is required for the operation of complex IV (citrochrome C oxidase) of the electron transport chain. Cardiolipin is found almost exclusively in the inner mitochondrial membrane. With age, there is a decrease in the number of cardiolipin. There is a theory that the loss of cardiolipin function is associated with the replacement of saturated fatty acids in its molecule with polyunsaturated fatty acids. To address this issue, it is necessary to introduce into the diet saturated fats, rich in primarily stearic fatty acid.
To increase the efficiency of delivery of saturated fatty acids into the membrane, the use of carriers is possible. For example, the use of saturated phosphatidylcholine (dipalmitrophosphatidylcholine and diseroylphosphatidylcholine), which, potentially, will be able to deliver saturated FA directly to cardiolipin [10]. Choline, as a carrier, easily passes through the cytosol and enters the mitochondria.
Phosphatidic acid (RA) inhibits mitochondrial division and stimulates the fusion of external membranes (Fig. 7).
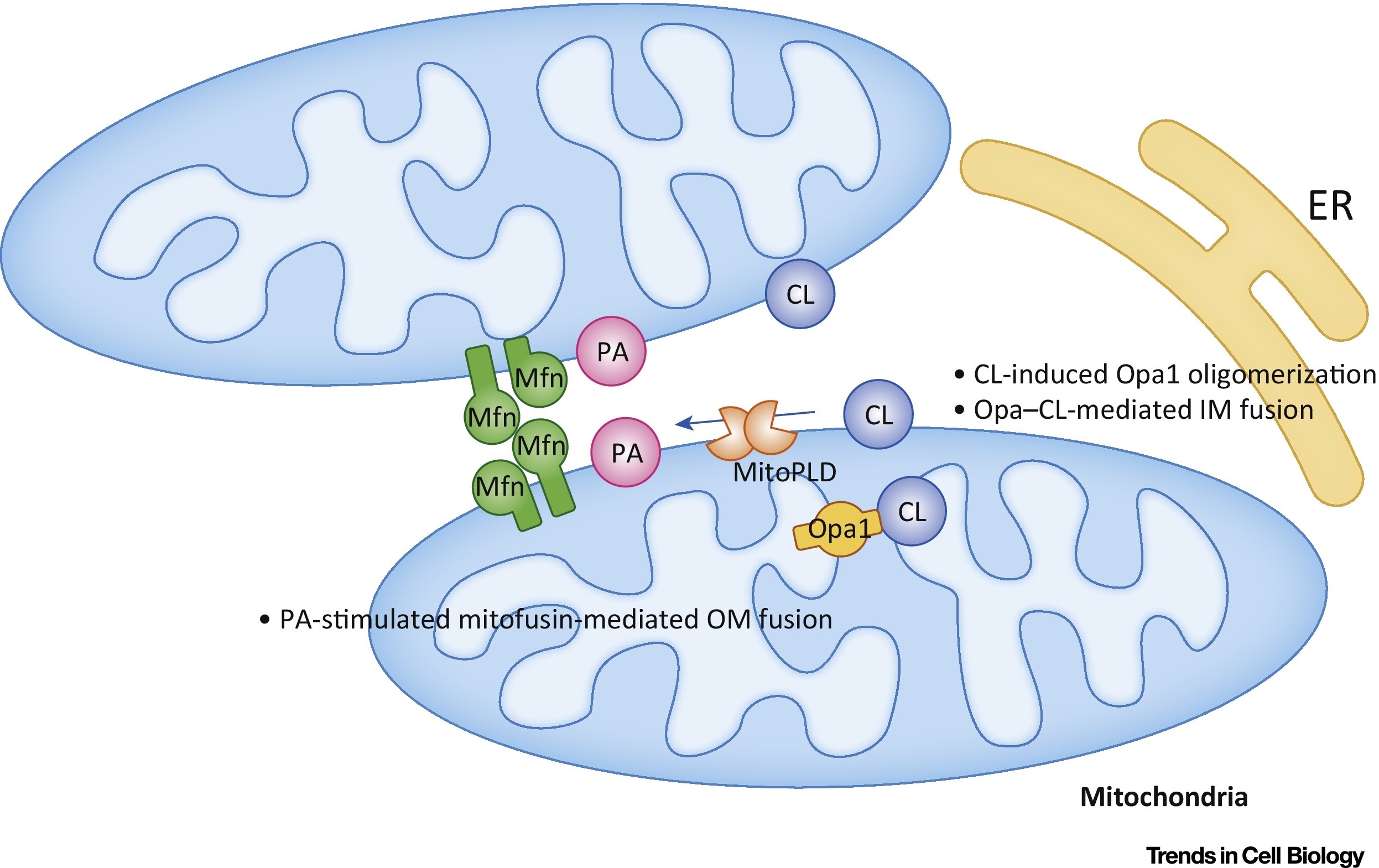
Fig.7 Regulation of mitochondrial phosphatidic acid (PA) and cardiolipin (CL) fusion [out of 9].
In the outer membrane (OM), RA stimulates mitofuzin-mediated (Mfn) fusion. In the inner membrane (IM), CL stimulates Opa1-mediated fusion. Abbreviations: ER - endoplasmic reticulum; MitoPLD, mitochondria-localized phospholipase D.
3. Regulation of the expression of mitofusins (proteins responsible for the dynamics of mitochondria)
Everything we talked about above (calorie restriction, stearic acid, phospholipids) affect the expression of mitofusins.
In addition, there are a number of drugs that can indirectly affect the dynamics of mitochondria. These include the use of metformin.
The most interesting is the use of substances that are able to directly affect the expression of mitofuzins. Leflunomide (leflunomide), which has been approved by the FDA [5,11], is one potential drug. It is an inducer of the expression of Mfn1 and Mfn2, and was registered as a drug for the treatment of rheumatoid arthritis.
Violation of the dynamics of mitochondria may be associated with impaired expression of proteins responsible for the fusion and division of mitochondria. In addition, the dysfunction of these proteins can be associated (and this happens most often) with their mutations. There are two approaches to the consideration of causal interactions of mitochondrial dysfunction.
Earlier it was believed that lifestyle, including overeating, leads to the formation of free radicals, oxidative stress, mutations of the mitochondrial genome and, consequently, impairment of mitochondrial function. However, recently there is convincing evidence that mitochondrial DNA mutations are inevitable, everyone has (heteroplasmic DNA point mutations) and is associated with replication errors, and not oxidative damage, to which mitochondrial DNA is fairly resistant [12]. Already at the stage of the fertilized egg, part of our mitochondria carry mutations. Over time, they divide, mutant mitochondria become larger, they can not properly perform their function.
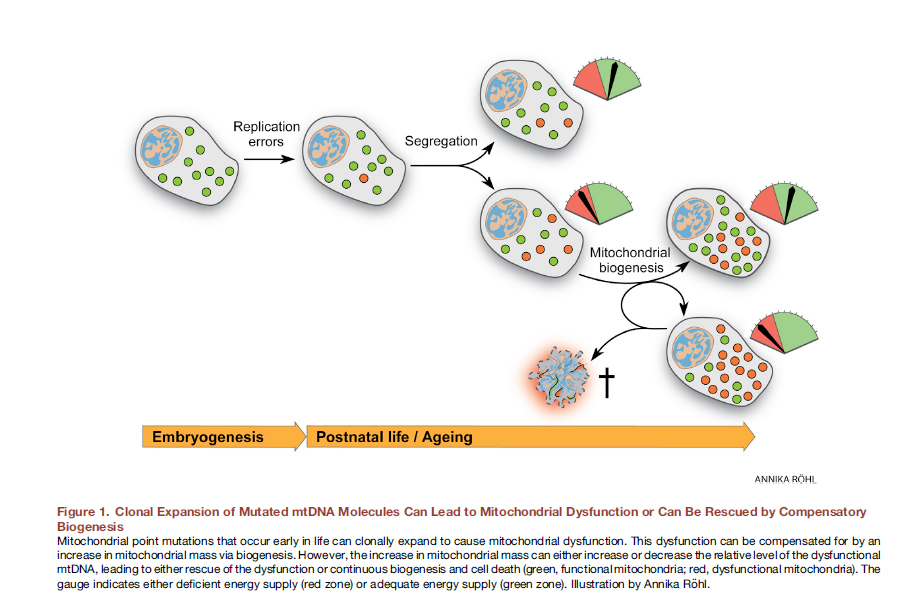
Fig. eightClonal expansion of mutated mtDNA molecules can lead to mitochondrial dysfunction or can be “saved” by compensatory biogenesis [out of 12].
Here it would be very useful to use the editing of the mitochondrial genome in vivo. It was shown that for mice with heteroplasmic DNA point mutations, considerable success has already been achieved with the help of the targeted zinc-finger nucleases (mtZFN) with delivery using an adenoviral vector [13].
Another promising method for eliminating mitochondrial dysfunction is mitochondrial transplantation. The essence of this approach is to “replace” damaged mitochondria with healthy exogenous mitochondria. For the first time, this approach was used clinically in children with myocardial ischemia. Autologous isolated mitochondria were used for transplantation, which were isolated from the rectus abdominis muscle (a biopsy was made, and then the preparation was prepared), and then injected by direct injection [14]. Various approaches to the introduction of mitochondria are being worked out: direct injection of isolated mitochondria (local administration) and systemic introduction into the bloodstream, when the mitochondria itself “seeks” in which cell to go to it. Groups of researchers are exploring the possibility of mitochondrial transplantation in Parkinson's disease, liver ischemia, stroke, mitochondrial diseases [15].
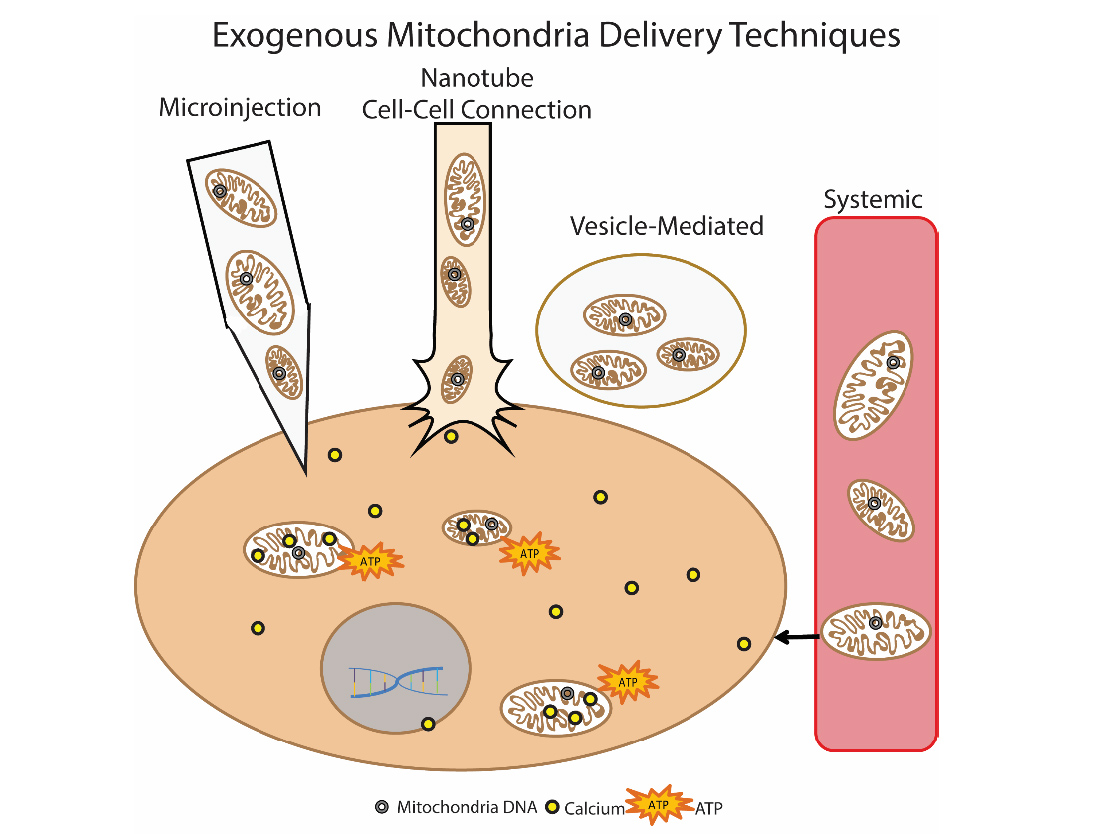
Fig.9. Methods of delivery of exogenous mitochondria into the cell
Author Olga Borisova
If you think that the most important story of living together for us begins during a wedding, then this is not at all the case. The most important story of the joint life of each person began more than a billion years ago, when our distant unicellular ancestors were forced to sign a “marriage contract” with those whom we now call mitochondria (see the theory of symbiogenesis).
Mitochondria have two membranes (internal and external) and their own hereditary material in the form of DNA (Fig. 1). On the inner membrane of the mitochondria is a system of oxidative phosphorylation, which provides the oxidation of energy substrates with the formation of ATP.
Fig. 1 . The schematic structure of the mitochondria.
In the marriage contract of the cell and mitochondria there is no point “in sickness and health,” and that’s good. If the mitochondria becomes old, the cell can kill it in the process of mitophagy, and the mitochondria, in turn, regulate the process of apoptosis in incapable and old cells. If the process of mutual quality control is violated, the mechanisms of aging are triggered. The mechanisms of apoptosis are violated, the number of free radicals that are not controlled by the mitochondria increases. This causes systemic inflammation, cell DNA damage. Thus, there is a strong relationship between MX dysfunction, age-related diseases, aging of the body and metabolic dysfunctions [1]. Metabolic dysfunction - the constant rider of the apocalypse of aging.
"Like a squirrel in a wheel" - the dynamics of mitochondria
Not all the blame for metabolic disorders lies with our overeating. Metabolic disorders are associated primarily with the inability of mitochondria to cope with nutrients. Mitochondria in a cell is not easy. We “feed” our cells either too much, then too little, and we present them with a “request” to give out energy in the form of ATP, the amount of which must exactly correspond to our needs. In order to “wriggle out” from this situation regularly, the mitochondria really use some “movements” - fission and fusion. These "mitodizations" unite under the name "dynamics of mitochondria". The balance between mitochondrial division and fusion is the central mechanism of bioenergetic adaptation to the metabolic needs of the cell [2, 3].
Most of all mitochondria are found in tissues with high energy needs - muscles, liver, brown adipose tissue, brain. It is not surprising that the dynamics of mitochondria in these tissues is better studied.
So, if a cell of any of these tissues (except for some neurons in the brain, about this later) receives a large amount of nutrients (intake exceeds costs), then the mitochondria are in a divided (fragmented) state. If the cell is in a state of hunger (revenues are less costly), then mitochondrial fusion occurs and they are in the United state. [3,4]. This is how cell homeostasis is maintained (Fig. 2).
Fig. 2 Regulation of morphology and bioenergetic efficiency of mitochondria in response to excessive or insufficient nutrient intake [of 2]
Cellular metabolic homeostasis depends on the balance between the consumption of nutrients and their consumption. Changes in nutrient supply leads to cellular adaptations to restore balance. Excess nutrition leads to fragmentation of the mitochondrial network, which causes a decrease in the bioenergy efficiency of mitochondria. This will avoid energy loss. In contrast, with metabolic hunger, mitochondria are lengthened to increase their bioenergetic efficiency.
What is the trick of these movements? If the cell is in a state of starvation, the mitochondrial fusion allows increasing their bioenergetic efficiency (the amount of ATP that is created per nutrient molecule). If an excess of nutrients enters the cell, then they can either be 1) stored or 2) this energy can be dissipated as heat. The task of mitochondria in this case is to dissipate more energy in the form of heat, to store less in the form of ATP (accumulation of NADH and ROS will lead to oxidative stress). Fragmentation of mitochondria allows them to reduce bioenergetic efficiency, the main mechanism for reducing which is considered "leakage" of protons.
So, we go to work, and the life of the mitochondria constantly proceeds in the division and fusion cycle mode (Figure 3).
Pic.3The balance of energy consumption and energy is associated with the corresponding changes in mitochondrial architecture and their bioenergy efficiency [of 3]
Physiological processes associated with an increase in energy demand and reduced energy supplies (for example, acute stress, starvation and the G1 / S phase) are characterized by elongation of mitochondria and respiration associated with the synthesis of ATP. On the other hand, the physiological processes associated with a decrease in energy demand and an increase in its supply (high nutrient levels, obesity, and type 2 diabetes) are associated with mitochondrial fragmentation, heat release, or a decrease in mitochondrial function.
Healthy division and fusion cycles are key to metabolic cell health
The normal cycle of mitochondrial division and their fusion is a key element in their quality control. Why? When mitochondrial division occurs, two daughter cells form, one of which has a higher membrane potential and goes further into the fusion-division cycle, while the other, with a more depolarized membrane, remains separated until the membrane potential is restored. If the potential is restored, it is reunited with the mitochondrial network. If it remains depolarized, then it is eliminated in the process of autophagy, which is the key to the quality of the mitochondrial pool (Fig. 4).
Long-term inhibition of mitochondrial division (with prolonged cell starvation) leads to the accumulation of damaged mitochondria, which cannot be segregated [3, 4].
On the other hand, an excess of nutrients leads to inhibition of mitochondrial fusion, which leads to disruption of the cycle of mitochondrial dynamics, increases intracellular mitochondrial heterogeneity. Yes, with an excess of food, mitochondrial fragmentation is protective, but long-term fragmentation, like long-term fusion, is detrimental to the quality control of mitochondria. There is no selective removal, the mitochondrial mass will decrease and consist of small depolarized mitochondria.
Fig.4. The life cycle of mitochondria and its regulation by the availability of nutrients [of 3]
Mitofuziny - not just some proteins
At the molecular level, mitochondrial fusion is a two-step process that requires a coordinated fusion of the outer and inner membranes during individual successive events. In mammals, this process is regulated by three proteins that belong to GTPases: Mfn1 and Mfn2 are necessary for the fusion of the outer membrane, and OPA1 for the fusion of the inner membrane. Other proteins are needed for division, Fis1 and Drp1.
The role of mitofusin proteins has been studied in loss-and gain-of function studies. Mice mutant for mitofusin proteins die even in mid-gestation, because in them the fusion of mitochondria becomes impossible. Mitofuzins are important for autophagy and mitophagy. Reducing the expression of Mfn2 in cardiomyocytes blocks the start of the autophagy process, because the fusion of autophagosomes with lysosomes is blocked. The depletion of Mfn2 leads to a decrease in the potential of mitochondrial membranes, to compensate, the respiratory chain decreases, glucose uptake increases and glycogen synthesis decreases. The cell switches to anaerobic glyoclycine, and this is the path to oncological cell degeneration. A deficiency of Mfn2 leads to neurodegenerative changes. Increased expression of Mfn2 in skeletal muscle increases their insulin sensitivity.
Mfn1 performs similar functions, however, probably in other tissues (expression of Mfn2 and Mfn1 differs in different tissues) - Mfn1 is more expressed in the heart, liver, pancreas, testicles, and Mfn2 in the heart, skeletal muscles, brain, brown adipose tissue .
Thus mitofusins are key regulators of mitochondrial dynamics. The expression of mitofusins is different in different organs, they provide bioenergetic efficiency and adaptation mechanisms to the availability of nutrients, and the “fate” of the cell depends on them. Not surprisingly, mitochondrial fusion proteins are potential targets for pharmacological interventions [2, 5].
Hypothalamus, mitochondria, metabolic dysfunction and aging
The dynamics of mitochondria is important in all cells. In the beta cells of the pancreas, mitochondria are nutrient sensors and signal generators of insulin synthesis, in muscles, the dynamics of mitochondria are important for the regulation of glucose metabolism, etc. However, a person is not just a collection of cells of a different type, each of which makes independent decisions. An organism is a system that has a central regulatory link for maintaining energy and glucose homeostasis. This major regulator is the hypothalamus.
The hypothalamus is located in the diencephalon and it is it that provides the interconnection of the nervous and humoral regulation systems. The hypothalamus neurons perceive, process and respond to signals from adipose tissue (leptin), pancreas (insulin), and other hormonal stimuli (ghrelin, cholecystokinin, pancreatic polypeptide, etc.). The hypothalamus controls the activity of the human endocrine system due to the fact that its neurons are able to secrete neuroendocrine transmitters that stimulate or suppress the production of hormones by the pituitary gland. In other words, the hypothalamus, whose mass does not exceed 5% of the brain, is the center of regulation of endocrine functions and maintaining homeostasis of the whole organism.
Even Dilman (Dilman V. M "Great biological clock") pointed to the leading role of the hypothalamus in the systematic development of metabolic dysfunction, leading to obesity, diabetes, cardiovascular diseases, cancer and aging. According to Dilman’s theory of hyper-adaptation, the sensitivity of the hypothalamic receptors to signals from the body’s tissues (leptin, insulin, etc.) gradually decreases gradually with age. In order to evoke his “answer” you need more and more of this or that hormone, more insulin, more leptin. Insulin and leptin resistance, metabolic diseases leading to aging and death are developing.
Depending on the functions performed, groups of neurons are combined into hypothalamic nuclei. One of them - arcuate (arcuate) core is a key regulator of eating behavior and metabolism. It can form orexigenic neuropeptides (stimulate appetite) and anorexigenic (suppress appetite), related, respectively, to AgRP and POMC neurons. Peripheral signals (insulin, ghrelin, leptin, etc.) affect the expression of peptides that stimulate or suppress the appetite, which ensures coherence of the central regulation (Fig. 5).
Fig. five.Hypothalamic control of energy metmbolism. The brain integrates metabolic signals (leptin, insulin, ghrelin, PYY3-36) from peripheral tissues, such as pancreas, adipose tissue, stomach. In the brain, specialized neural networks coordinate adaptive changes in the absorption and consumption of food [of 5].
So who and how regulates the sensitivity of the hypothalamic neurons?
The study of the dynamics of mitochondria in the brain tissues showed that the dynamics of mitochondria plays a significant role in the ability of the hypothalamus neurons to control the level of glucose and energy homeostasis in the body [6,7,8].
In AgRP neurons (hunger-promoting AgRP neurons), which stimulate appetite and regulate weight gain, starvation leads to the division of mitochondria, and high-fat feeding leads to fusion. That is, the mitochondrial response is different from that in most other cells.
The fusion of MX in these neurons regulates the electrical activity in response to a high-fat diet, stimulating the production of orexigenic peptide (AgRP peptide); it is necessary for weight gain and fat deposition with an excess of nutrients. Deletions of Mfn1 and Mfn2 in these neurons resulted in less weight gain in rats by reducing the level of circulating leptin.
POMC neurons (suppress appetite) have the opposite function, and the dynamics of mitochondria in response to the intake of nutrients they have different. Reduced expression of mitofusins in these neurons leads to disruption of the association of mitochondria with EPS, and, as a result, hyperphagia, leptin resistance and obesity. At the same time, food consumption increased, while energy consumption decreased.
Thus, the body's response to a high-fat diet depends on the patterns of mitochondrial dynamics in the hypothalamus neurons. Remodeling of mitochondria in neurons provides their response to the intake of nutrients into the body, stimulates the production of neuropeptides that will either stimulate or suppress appetite, affecting metabolism at the level of the organism (Fig.6).
Fig.6. Metabolic adaptation to environmental stimuli [of 2]
In response to exogenous stimuli, Mfns are involved in the transduction of metabolic signaling in various organs, which ensures the maintenance of homeostasis of the whole body's energy. In particular, in response to food intake, temperature changes, stress or exercise, brown adipose tissue, the brain, heart or skeletal muscles adapt their metabolism to control nutrition, body weight, contractile functions, antioxidant response or insulin sensitivity.
How to influence the dynamics of mitochondria?
1. Nutrition and Exercise
Nutrition Cycles Excess food and a high-fat diet (HFD) inhibit mitochondrial fusion in cells (in some neurons of the brain, the mechanism is different). An incomplete mitochondrial division-fusion cycle disrupts autophagy → the intracellular heterogeneity of mitochondria increases → no selective removal of mitochondria occurs → the mitochondria with dysfunction accumulate.
Calorie restriction (fed / fasting cycle) stimulates bioenergetic adaptation, ensuring the functioning of mitochondrial quality mechanisms.
2. Healthy membranes: stearic acid, cardiolipin, phosphatidic acid
All key processes depend on the “health” of mitochondrial membranes — autophagy, mitophagy, apoptosis, the association of mitochondria with the endoplasmic reticulum, and the dynamics of mitochondria. Cell membranes are composed of lipids and proteins. The remodeling of these membranes is controlled by the interactions between specific lipids and proteins.
Palmitic (C16) and stearic (C18) are among the saturated fatty acids. Stearic acid (C18: 0) has been shown to stimulate mitochondrial fusion. Its action is associated with the effect on mitofuzina. In mice, dietary supplements of stearic acid can partially restore mitochondrial dysfunction caused by mutations in the Pink1 or parkin genes. In the neutrophils of people who are on a low-C18: 0 diet for 2 days, the mitochondria are in a fragmented state (50% of the cells had fragmented MX, 10% united MX). Their use of stearic acid led to mitochondrial fusion after 3 hours [8]. Thus, steric acid is important for maintaining cycles of mitochondrial dynamics. Most of the stearic acid is in cocoa beans (31-34%).
Phospholipids are the main components of organelle membranes. They also regulate the dynamics of mitochondria, and their influence is different [9].
Cardiolipin (CL) stimulates mitochondrial division and fusion of internal membranes.
Cardiolipin is required for the operation of complex IV (citrochrome C oxidase) of the electron transport chain. Cardiolipin is found almost exclusively in the inner mitochondrial membrane. With age, there is a decrease in the number of cardiolipin. There is a theory that the loss of cardiolipin function is associated with the replacement of saturated fatty acids in its molecule with polyunsaturated fatty acids. To address this issue, it is necessary to introduce into the diet saturated fats, rich in primarily stearic fatty acid.
To increase the efficiency of delivery of saturated fatty acids into the membrane, the use of carriers is possible. For example, the use of saturated phosphatidylcholine (dipalmitrophosphatidylcholine and diseroylphosphatidylcholine), which, potentially, will be able to deliver saturated FA directly to cardiolipin [10]. Choline, as a carrier, easily passes through the cytosol and enters the mitochondria.
Phosphatidic acid (RA) inhibits mitochondrial division and stimulates the fusion of external membranes (Fig. 7).
Fig.7 Regulation of mitochondrial phosphatidic acid (PA) and cardiolipin (CL) fusion [out of 9].
In the outer membrane (OM), RA stimulates mitofuzin-mediated (Mfn) fusion. In the inner membrane (IM), CL stimulates Opa1-mediated fusion. Abbreviations: ER - endoplasmic reticulum; MitoPLD, mitochondria-localized phospholipase D.
3. Regulation of the expression of mitofusins (proteins responsible for the dynamics of mitochondria)
Everything we talked about above (calorie restriction, stearic acid, phospholipids) affect the expression of mitofusins.
In addition, there are a number of drugs that can indirectly affect the dynamics of mitochondria. These include the use of metformin.
The most interesting is the use of substances that are able to directly affect the expression of mitofuzins. Leflunomide (leflunomide), which has been approved by the FDA [5,11], is one potential drug. It is an inducer of the expression of Mfn1 and Mfn2, and was registered as a drug for the treatment of rheumatoid arthritis.
Mitochondrial Gene Therapy
Violation of the dynamics of mitochondria may be associated with impaired expression of proteins responsible for the fusion and division of mitochondria. In addition, the dysfunction of these proteins can be associated (and this happens most often) with their mutations. There are two approaches to the consideration of causal interactions of mitochondrial dysfunction.
Earlier it was believed that lifestyle, including overeating, leads to the formation of free radicals, oxidative stress, mutations of the mitochondrial genome and, consequently, impairment of mitochondrial function. However, recently there is convincing evidence that mitochondrial DNA mutations are inevitable, everyone has (heteroplasmic DNA point mutations) and is associated with replication errors, and not oxidative damage, to which mitochondrial DNA is fairly resistant [12]. Already at the stage of the fertilized egg, part of our mitochondria carry mutations. Over time, they divide, mutant mitochondria become larger, they can not properly perform their function.
Fig. eightClonal expansion of mutated mtDNA molecules can lead to mitochondrial dysfunction or can be “saved” by compensatory biogenesis [out of 12].
Here it would be very useful to use the editing of the mitochondrial genome in vivo. It was shown that for mice with heteroplasmic DNA point mutations, considerable success has already been achieved with the help of the targeted zinc-finger nucleases (mtZFN) with delivery using an adenoviral vector [13].
Mitochondrial transfer
Another promising method for eliminating mitochondrial dysfunction is mitochondrial transplantation. The essence of this approach is to “replace” damaged mitochondria with healthy exogenous mitochondria. For the first time, this approach was used clinically in children with myocardial ischemia. Autologous isolated mitochondria were used for transplantation, which were isolated from the rectus abdominis muscle (a biopsy was made, and then the preparation was prepared), and then injected by direct injection [14]. Various approaches to the introduction of mitochondria are being worked out: direct injection of isolated mitochondria (local administration) and systemic introduction into the bloodstream, when the mitochondria itself “seeks” in which cell to go to it. Groups of researchers are exploring the possibility of mitochondrial transplantation in Parkinson's disease, liver ischemia, stroke, mitochondrial diseases [15].
Fig.9. Methods of delivery of exogenous mitochondria into the cell
Author Olga Borisova
Literature
1. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. «Mammalian mitochondria and aging: an update.» Cell metabolism 25.1 (2017): 57-71.
www.sciencedirect.com/science/article/pii/S1550413116305022
2. Schrepfer, Emilie, and Luca Scorrano. «Mitofusins, from mitochondria to metabolism.» Molecular cell 61.5 (2016): 683-694.
www.sciencedirect.com/science/article/pii/S1097276516001337#fig1
3. Marc Liesa, Orian Shirihai “Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure” Cell methabolism (2013): 491-506
www.sciencedirect.com/science/article/pii/S1550413113001046#fig3
4. Ramos, Eduardo Silva, Nils-Göran Larsson, and Arnaud Mourier. «Bioenergetic roles of mitochondrial fusion.» Biochimica et Biophysica Acta (BBA)-Bioenergetics 1857.8 (2016): 1277-1283.
www.sciencedirect.com/science/article/pii/S0005272816300858
5. Cunarro, Juan, et al. «Hypothalamic mitochondrial dysfunction as a target in obesity and metabolic disease.» Frontiers in endocrinology 9 (2018): 283.
www.frontiersin.org/articles/10.3389/fendo.2018.00283/full
6. Marcelo O.Dietrich et al. «Mitochondrial Dynamics Controlled by Mitofusins Regulate Agrp Neuronal Activity and Diet-Induced Obesity”.
www.sciencedirect.com/science/article/pii/S0092867413010957#figs2
7. Steculorum, Sophie M., and Jens C. Brüning. „Sweet mitochondrial dynamics in VMH neurons.“ Cell metabolism 23.4 (2016): 577-579.
www.sciencedirect.com/science/article/pii/S1550413116301176
8. Senyilmaz-Tiebe, Deniz, et al. „Dietary stearic acid regulates mitochondria in vivo in humans.“ Nature communications 9.1 (2018): 3129.
www.nature.com/articles/s41467-018-05614-6
9. Kameoka, Shoichiro, et al. „Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics.“ Trends in cell biology (2017).
www.sciencedirect.com/science/article/pii/S0962892417301587
10. raypeatforum.com/community/threads/mitolipin-liquid-saturated-phosphatidylcholine-pc-mix.10398
11. Miret-Casals, Laia, et al. „Identification of new activators of mitochondrial fusion reveals a link between mitochondrial morphology and pyrimidine metabolism.“ Cell chemical biology25.3 (2018): 268-278.
12. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. „Mammalian mitochondria and aging: an update.“ Cell metabolism 25.1 (2017): 57-71.
13. Gammage et al. “Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo” Nature medicine, 2017
www.nature.com/articles/s41591-018-0165-9
14. Emani, Sitaram M., et al. „Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury.“ The Journal of thoracic and cardiovascular surgery 154.1 (2017): 286-289.
www.jtcvs.org/article/S0022-5223(17)30258-1/fulltext
15. McCully, James D., et al. „Mitochondrial transplantation: From animal models to clinical use in humans.“ Mitochondrion 34 (2017): 127-134.
www.sciencedirect.com/science/article/pii/S1567724917300053
www.sciencedirect.com/science/article/pii/S1550413116305022
2. Schrepfer, Emilie, and Luca Scorrano. «Mitofusins, from mitochondria to metabolism.» Molecular cell 61.5 (2016): 683-694.
www.sciencedirect.com/science/article/pii/S1097276516001337#fig1
3. Marc Liesa, Orian Shirihai “Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure” Cell methabolism (2013): 491-506
www.sciencedirect.com/science/article/pii/S1550413113001046#fig3
4. Ramos, Eduardo Silva, Nils-Göran Larsson, and Arnaud Mourier. «Bioenergetic roles of mitochondrial fusion.» Biochimica et Biophysica Acta (BBA)-Bioenergetics 1857.8 (2016): 1277-1283.
www.sciencedirect.com/science/article/pii/S0005272816300858
5. Cunarro, Juan, et al. «Hypothalamic mitochondrial dysfunction as a target in obesity and metabolic disease.» Frontiers in endocrinology 9 (2018): 283.
www.frontiersin.org/articles/10.3389/fendo.2018.00283/full
6. Marcelo O.Dietrich et al. «Mitochondrial Dynamics Controlled by Mitofusins Regulate Agrp Neuronal Activity and Diet-Induced Obesity”.
www.sciencedirect.com/science/article/pii/S0092867413010957#figs2
7. Steculorum, Sophie M., and Jens C. Brüning. „Sweet mitochondrial dynamics in VMH neurons.“ Cell metabolism 23.4 (2016): 577-579.
www.sciencedirect.com/science/article/pii/S1550413116301176
8. Senyilmaz-Tiebe, Deniz, et al. „Dietary stearic acid regulates mitochondria in vivo in humans.“ Nature communications 9.1 (2018): 3129.
www.nature.com/articles/s41467-018-05614-6
9. Kameoka, Shoichiro, et al. „Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics.“ Trends in cell biology (2017).
www.sciencedirect.com/science/article/pii/S0962892417301587
10. raypeatforum.com/community/threads/mitolipin-liquid-saturated-phosphatidylcholine-pc-mix.10398
11. Miret-Casals, Laia, et al. „Identification of new activators of mitochondrial fusion reveals a link between mitochondrial morphology and pyrimidine metabolism.“ Cell chemical biology25.3 (2018): 268-278.
12. Kauppila, Timo ES, Johanna HK Kauppila, and Nils-Göran Larsson. „Mammalian mitochondria and aging: an update.“ Cell metabolism 25.1 (2017): 57-71.
13. Gammage et al. “Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo” Nature medicine, 2017
www.nature.com/articles/s41591-018-0165-9
14. Emani, Sitaram M., et al. „Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury.“ The Journal of thoracic and cardiovascular surgery 154.1 (2017): 286-289.
www.jtcvs.org/article/S0022-5223(17)30258-1/fulltext
15. McCully, James D., et al. „Mitochondrial transplantation: From animal models to clinical use in humans.“ Mitochondrion 34 (2017): 127-134.
www.sciencedirect.com/science/article/pii/S1567724917300053