Comparison of biological sequences
The HABR tetrapeptide is incomparable
. Comparison of sequences of characters is a seemingly simple matter, but applied to biology, almost out of the blue it encounters a bunch of problems. Some tasks of modern biology cannot be solved at the present stage in the development of computer technology. In this article, I will show on fingers what is so special about biological sequences and why they need special algorithms. Biologists and especially bioinformatics are not recommended to read - there is a risk of dying from boredom.
Biological sequences are the primary structure of biological macromolecules. Namely, proteins and DNA / RNA. (There are also carbohydrates, for example, starch, but they consist of the same monomers and therefore are not interesting.) The DNA sequence determines the protein sequence, the protein sequence determines its spatial structure, the structure determines the functions of the protein, and the totality of the functions of different proteins is called life. It is the differences in the functioning of different proteins that we, in essence, differ from each other. Molecules can be compared, roughly speaking, for two reasons:
1) by comparing the proteins of different organisms in pairs, we can say which organisms are more similar to each other and which less;
2) comparing a dozen or two proteins at the same time, we can find structurally and functionally important regions (they are usually identical in related organisms), which is useful when creating artificial proteins (drug design, nanotechnology, in the good sense of the word).
It’s better to compare proteins, rather than DNA, if only because the “alphabet” has more proteins (20 amino acids versus 4 nucleotides), and the chance of chance coincidence is lower, so the discussion will be about proteins. Generally speaking, the structure should be compared, not the sequence (since we still do not quite know how the second defines the first); but it so happened that we learned to figure out the sequences well, and obtaining a spatial structure is a very time-consuming task and, in the opinion of many, is more likely an art. Therefore, we have to compare the sequence. Comparison of sequences of biological macromolecules is called alignment - the lines are written one under the other in such a way as to achieve coincidence or similarity of characters in the largest number of positions. One amino acid residue in a protein corresponds toone letter of the latin alphabet in sequence.
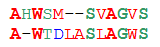
Taking into account the degree of similarity of monomers
We are accustomed to consider the two sequences the more similar, the more characters in them coincide. In biology, this approach is no good. Consider two pairs of sequences:
Пара 1 Пара 2ASDLV ASDLVATEIV AWDKVOld Hamming unequivocally says that the second alignment is better: in the first pair, only two out of five characters coincide, and in the second - three. But let's look at their structures:
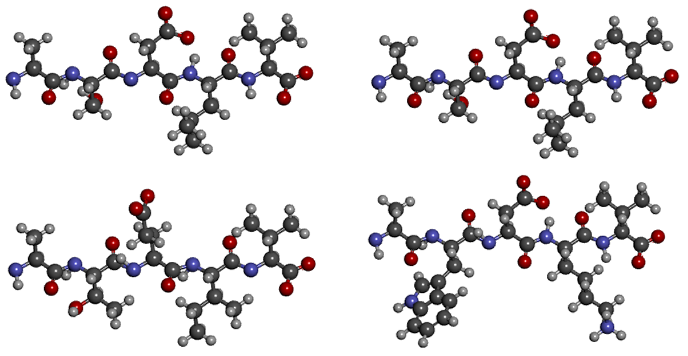
S and T, D and E — those residues by which the sequences of the first pair differ — differ in structure by exactly one carbon atom, L and I are generally isomers. In most cases, such a difference is not reflected in the structure of the protein. But the replacement of small polar serine with large non-polar tryptophan or hydrophobic leucine with charged lysine, as in the second pair, will greatly change the structure of the molecule. Thus, in the first alignment, the sequences are almost identical, and in the second, they are very different.

Taking into account the structural features of the characters being compared is quite simple: the so-called replacement matrix, like the one in the figure. The larger the number at the intersection of the matching letters, the greater the quantitative similarity indicator (called “Score”, or “Score”).
A S D L V A S D L V A T E I V A W D K VScore = 4+1+2+2+4 = 13 Score = 4-3+6-2+4 = 9Matrices are compiled based on statistics of amino acid substitutions in proteins with already known structure: the more often one letter is replaced by another, the greater the number at their intersection. The problem is that periodically new matrices appear, better than the previous ones; accordingly, they are all somewhat bad and do not take into account something.
Accounting for insertions and deletions
I don’t know how others, but at one time it struck me how similar the proteins of different organisms are. Where is the man - and where is the rabbit, and serum albumin in both of them differ by only a few amino acids. But they are still somewhat different: mutational variability is an integral property of the living. In fact, evaluating the similarity of proteins, we estimate the number of mutations that have occurred on the path of evolutionary transformation of one protein into another. Consideration of mutations of the first type - substitutions - we have considered above. Two more types - insertions and deletions (deletions) - we will consider now.
Insertions and deletions in bioinformatics are considered as one entity for a simple reason - having two sequences, one of which has an extra letter, we cannot determine which of the sequences is “original”. The hypothetical common ancestor of both organisms died out millions of years ago, so we do not know for sure whether insertion or deletion takes place in this case. For this reason, the term "inde" appeared (insertion + deletion); in Russian bioinformatics, the term "deletion" or tracing paper from the English "gap" is more common.

Two possible variants of the origin of related sequences. A pair of sequences from the bottom line are really existing “molecules”, at the top there is a hypothetical ancestor that could be either on the left or on the right (or in general neither way).
If there are deletions in the alignment, they are fined. The more deletions, the more points are subtracted from the account. And again, consider two alignments:
ASGHDLV AMSDCLVAT--EIV A-TD-VVIn both alignments, two amino acids "fell out". In both cases, formally, two mutations occurred. In fact, in the first case, one mutation occurred . Two remainders long. But one. One evolutionary event. And its probability is greater than two separate ones, as in the second example. Therefore, the alignment score on the left should be higher, and the first two sequences should be considered more similar.
To account for deletions, the so-called affine fine consisting of a fine “for opening” and “for continuing” a deletion. The first is usually an order of magnitude or two more than the second (about 10 and 1, respectively, or 10 and 0.1). In the example on the left, we have one discovery and one continuation, on the right - two discoveries.
The problem of accounting for deletions is not limited to this. Depending on the location of the deletion in the molecule, it can affect the structure to a greater or lesser extent. Some algorithms, such as Clustal, take into account “residual-specific penalties,” some do not. Usually this boils down to the fact that deletions in hydrophilic regions - potential loops dangling in an aqueous environment - are fined less than in hydrophobic (potential tightly packed globule nuclei). The question is, how much should these penalties differ, and only if hydropathy should serve as a criterion for differences, apparently, everyone decides for himself ...
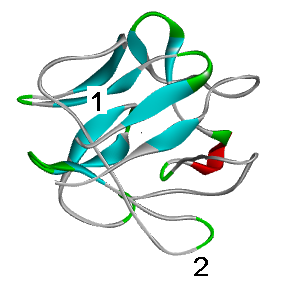
The mutation in site No. 1 — a tightly packed and strictly structured (beta-strand) core of the globule — can greatly disrupt the protein structure. Mutation in plot No. 2 (unstructured relatively labile loop), most likely, will not affect it in any way.
Alignment Calculation
So far, we have been considering evaluating the finished alignment without wondering how it turns out. The average protein consists of several hundred amino acids, it’s obviously impossible to align such long lines “by eye”. Although there are different ways to obtain alignments, the Needleman-Wunsch algorithm and its modifications are currently considered optimal . I will omit the essence of the algorithm: the mathematical audience can easily figure it out on their own, the rest
In the beginning, I said that aligning a dozen or two proteins at the same time, we can find structurally and functionally important sites in them. For example, the amino acids in the figure that make up the active center of trypsin-like proteases clearly show themselves as being identical in 20 similar proteins (corresponding positions are indicated by asterisks).

Amino acids that determine the catalytic properties of the enzyme are conservative, i.e. do not change during evolution. The active center of the enzyme must correspond to the geometry and chemistry of the substrate accurate to the atom, so the mutation in it means the "end of the career" of the enzyme as a catalyst. In fact, of course, mutations occur anywhere in the molecule; but proteins with a mutation in the functional region make their host unviable, therefore, it seems to us that they are not.
In conditions when obtaining a full three-dimensional structure of a protein is still a luxury in a certain sense, the ability to find such patterns is very valuable. The ambush here is that we cannot get the mathematically optimal alignment of even a dozen proteins. If a two-dimensional array is used to align two proteins, ten-dimensional, respectively, are needed to align 10 proteins. Even if each sequence has a length of 100 characters (which corresponds to a very small protein), the dimension of the array is so huge (100 10 ) that a modern computer will count it for thousands of years. I will probably tell you about how biologists can still get out in this situation another time.