Interesting results on the evolutionary systematics of prokaryotes or “multi-species origin”
Phylogenetic taxonomy is trying to determine the kinship of various organisms and their evolutionary proximity. If not so long ago this was judged by the external signs of organisms (morphology, more precisely), now they have unambiguously passed on to judgment by comparing the genomes of these organisms.
But the body’s DNA consists of many nucleotides and it is very difficult to take into account all of them to determine the similarity of organisms. In addition, DNA is constantly evolving. Therefore, biologists began to rely on ribosomal ribonucleic acid (rRNA), because these molecules were found in all cellular life forms, their functions are associated with the most important translation process for the body, the primary structure as a whole is characterized by high conservatism.
It is believed that a feature of rRNA is that it is outside the scope of selection, therefore, these molecules evolve as a result of spontaneous mutations that occur at a constant rate, and the accumulation of such mutations depends only on time. Thus, a measure of the evolutionary distance between organisms is the number of nucleotide substitutions in the molecules of the compared rRNAs.
It is known that 3 types of rRNA are present in the ribosomes of prokaryotes and eukaryotes. The information capacity of large molecules is larger, but more difficult to analyze. Therefore, the most convenient was the analysis of medium-sized rRNA molecules: 16S (~ 1600 nucleotides). The systematics is based on the calculation of similarity coefficients of the compared organisms. It is on the basis of rRNA analysis that modern systematics distinguishes three domains of bacteria, archaea and eukaryotes, and the systematics, bacteria and archaea of the 10th edition of Bergi are also based on this.
Here is the current situation in this area. I made an attempt to create the basis for a slightly different, if you want an alternative, systematics. Why? The conservatism of rRNA is nevertheless not large enough, only some of its parts are conservative. And since there are quite variable parts in rRNA, we have to make assumptions and assume where there were breaks and insertions of individual fragments during mutation. A so-called alignment is now done with a very large error.
As a result, I came to the conclusion that when comparing genomic sequences, it is necessary to compare such regions that were not mutated at all, and which are absolutely identical in different organisms.
We look what came of it.
Are there DNA fragments that do not mutate at all for a long time?
But are there any sites that have not undergone mutations at all, and are absolutely identical in different organisms? It turns out there is. A number of proteins (their DNA code) are absolutely identical for many species assigned to the same genus, or even the family. But transport RNAs (tRNAs) are even more conservative. In the bacterial chromosome, as a rule, all 20 types of tRNA exist, each of which is responsible for transporting a specific amino acid to the site of protein synthesis. And on the basis of them it is possible to trace the evolutionary relationship not only of individual families, but also of entire classes and even types.
In this study, I relied on only one tRNA that transports alanine (Ala tRNA). Therefore, conclusions about the evolutionary connection do not claim the final result. To do this, compare the results for other tRNAs. Nevertheless, the study done allows us to outline a number of provisions on the proximity of certain genera of bacteria. Then it will be shown that close evolutionary connections cannot be discussed in terms of a phylogenetic tree. This is due to the fact that there is horizontal transfer and conjugation in bacteria, and other approaches are required.
Genome analysis
The focus will be on the genus Yersinia (Plague bacillus), but as a result of the study, others such as Shewanella, Pseudomonas, Vibrio, Erythrobacter, Pseudoalteromonas, Photobacterium, and a number of other (109 loci in total)
tRNAs in bacteria, as a rule, have a constant length 76 nucleotides, while the anticodon is located at positions 34.35.36. Alanine in DNA is encoded by four codons: GCT, GCC, GCA, GCG. Therefore, 4 types of Ala tRNA are potentially possible, with anticodons AGC, GGC, TGC, CGC.
But the vast majority of the bacteria in question in the genome have only 2 species Ala tRNA_GCA and Ala tRNA_GCC. There are, of course, exceptions.
For analysis, we used sequenced DNA genomes that are available in the NCBI database. All identical non-mutated tRNAs were labeled with a unique identifier (Id). With the help of a computer program written for analysis, in a semi-automatic mode with manual verification, a list of various types of Ala tRNA was compiled , and their location in a particular sequenced locus.
Results
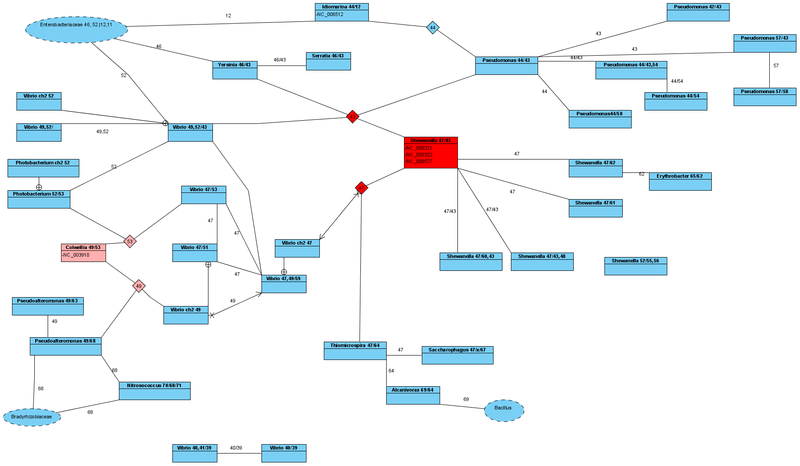
All examined strains of the genus Yersinia (9 pcs.) Have the same Ala tRNA_GCA with Id = 00046 and Ala tRNA_GCC with Id = 00043 in the genome. Based on this fact, we can conclude that indeed these strains have a strong evolutionary relationship, and therefore all of them belong to the genus Yersinia.
Now the genus Yersinia belongs to the Enterobacteriaceae family.But on the basis of the analysis done, within the framework of the Ala tRNA similarity, this relationship is unreasonable .
If you look at the classic representatives of the Enterobacteriaceae family, such as Escherichia, Salmonella, Shigella, Citrobacter, Cronobacter, Klebsiella, Pectobacterium, then they all have completely different Ala tRNAs. Namely, they have Ala tRNA_GCA with Id = 00011 and Ala tRNA_GCC with Id = 00012. On this basis, we can consider the listed genera of the Enterobacteriaceae family as classical representatives.
And only with the genus Photorhabdus, along the same line Ala tRNA_GCA with Id = 00046, Yersinia has a connection. Therefore, the genus Yersinia has traits from different families . We will call such a genus a transitional genus between different families .
So, if the Yersinia genus is associated with the Enterobacteriaceae family only by one configuration of the non-mutated Ala tRNA_GCA with Id = 00046 (and even partially), then the question arises of which family of the Yersinia genus is connected by another configuration of the non-mutated Ala tRNA_GCC with Id = 00043?
It turns out that the most direct relationship along this line is the genus Shewanella (family: Shewanellaceae, order: Alteromonadales, class: gamma-proteobacteria). At the same time, the Ala tRNA_GCC line connecting them with Id = 00043 is the key in evolutionary terms, since it is also present in the genera Pseudomonas and Vibrio. All these relations are closer than follows from the modern classification, where these genera are united only at the class level.
In turn, some representatives of the genus Shewanella have the same Ala tRNA_GCA with Id = 00047 and Ala tRNA_GCC with Id = 00043. Having already found out that Yersinia and Shewanella have a connection on one line (Id = 00043), it is interesting to whom the genus Shewanella is connected on another line (Id = 00047). It turns out that this line is also quite key in evolutionary terms. It branches further and Ala tRNA_GCA with Id = 00047 is also possessed by representatives of the genera Vibrio, Thiomicrospira, Saccharophagus.
We can also trace these connections further (see the figure). But now it’s clear that in order to systematize these evolutionary relations, it is necessary to slightly change the approach to their description.
higher resolution
Conclusions
It should be emphasized once again that all the conclusions made in this work are based on the analysis of only tRNA Ala, and of course, for recognition of the results, verification of other types of tRNA is needed. But nevertheless, now we can draw some conclusions and describe how to change approaches to taxonomy.
Of sufficient complexity is the determination of which species is more evolutionarily ancient and which is younger. But based on the hypothesis that the biological world has evolved from simple to complex, we have at least one indisputable fact. If a bacterium has two chromosomes, then it seems obvious that it is evolutionarily more young than having one.
Therefore, as part of our study, we can confidently say that the genus Vibrio is younger than Shewanella or Yersinia. And then, since Vibrio and Shewanella are united identical by Ala tRNAGCA with Id = 00047, then with a high probability, the genus Vibrio descended from Shewanella and so one chromosome originated. After which other representatives of the genus Vibrio descended from the genus Colwellia, and so the second chromosome occurred. As a result of combining these chromosomes in one organism, we can talk about the genus Vibrio, which descended from Shewanella along one line and Colwellia along the other.
Thus, we should not speak of descent from one ancestor, but about at least two, or even more.
With monochromosomal bacteria, it is more difficult to determine the direction of evolution (who is younger and who is older). But based on two-chromosomal species, we can say, since there is a Vibrio species with Ala tRNA_GCA with Id = 00049 and Ala tRNA_GCC with Id = 00043; and there are also Vibrio species with Ala tRNA_GCA with Id = 00047, then Ala tRNA_GCA with Id = 00047 and Ala tRNA_GCC with Id = 00043 originally existed. And they were contained in Shewanella, and therefore it must be recognized as the most ancient organism, and put in the basis of the taxonomy of the organisms considered here.
Then we can conclude that from Shewanella in one line came Yersinia. From Yersinia, in turn, Photorhabdus, from which the entire family Enterobacteriaceae originated. But this is only one line. According to another, we already mentioned what kind of birth descended from Shewanella.
The multi-species origin greatly confuses the evolutionary picture, but there is nothing to be done about it - such is the complexity of speciation, and we only need to most accurately reflect them in conditions where not all species are known.
upd. Oh yes. I completely forgot, then they started talking - well, why should programmers fool their heads with all kinds of biology. So I actually wanted to interest programmers with this topic, since they are the ones who are able to write algorithms for bio-calculations. I just don’t have the strength to conduct a more complete analysis. Suddenly someone will be interested in mercy, please write in a personal.
But the body’s DNA consists of many nucleotides and it is very difficult to take into account all of them to determine the similarity of organisms. In addition, DNA is constantly evolving. Therefore, biologists began to rely on ribosomal ribonucleic acid (rRNA), because these molecules were found in all cellular life forms, their functions are associated with the most important translation process for the body, the primary structure as a whole is characterized by high conservatism.
It is believed that a feature of rRNA is that it is outside the scope of selection, therefore, these molecules evolve as a result of spontaneous mutations that occur at a constant rate, and the accumulation of such mutations depends only on time. Thus, a measure of the evolutionary distance between organisms is the number of nucleotide substitutions in the molecules of the compared rRNAs.
It is known that 3 types of rRNA are present in the ribosomes of prokaryotes and eukaryotes. The information capacity of large molecules is larger, but more difficult to analyze. Therefore, the most convenient was the analysis of medium-sized rRNA molecules: 16S (~ 1600 nucleotides). The systematics is based on the calculation of similarity coefficients of the compared organisms. It is on the basis of rRNA analysis that modern systematics distinguishes three domains of bacteria, archaea and eukaryotes, and the systematics, bacteria and archaea of the 10th edition of Bergi are also based on this.
Here is the current situation in this area. I made an attempt to create the basis for a slightly different, if you want an alternative, systematics. Why? The conservatism of rRNA is nevertheless not large enough, only some of its parts are conservative. And since there are quite variable parts in rRNA, we have to make assumptions and assume where there were breaks and insertions of individual fragments during mutation. A so-called alignment is now done with a very large error.
As a result, I came to the conclusion that when comparing genomic sequences, it is necessary to compare such regions that were not mutated at all, and which are absolutely identical in different organisms.
We look what came of it.
Are there DNA fragments that do not mutate at all for a long time?
But are there any sites that have not undergone mutations at all, and are absolutely identical in different organisms? It turns out there is. A number of proteins (their DNA code) are absolutely identical for many species assigned to the same genus, or even the family. But transport RNAs (tRNAs) are even more conservative. In the bacterial chromosome, as a rule, all 20 types of tRNA exist, each of which is responsible for transporting a specific amino acid to the site of protein synthesis. And on the basis of them it is possible to trace the evolutionary relationship not only of individual families, but also of entire classes and even types.
In this study, I relied on only one tRNA that transports alanine (Ala tRNA). Therefore, conclusions about the evolutionary connection do not claim the final result. To do this, compare the results for other tRNAs. Nevertheless, the study done allows us to outline a number of provisions on the proximity of certain genera of bacteria. Then it will be shown that close evolutionary connections cannot be discussed in terms of a phylogenetic tree. This is due to the fact that there is horizontal transfer and conjugation in bacteria, and other approaches are required.
Genome analysis
The focus will be on the genus Yersinia (Plague bacillus), but as a result of the study, others such as Shewanella, Pseudomonas, Vibrio, Erythrobacter, Pseudoalteromonas, Photobacterium, and a number of other (109 loci in total)
tRNAs in bacteria, as a rule, have a constant length 76 nucleotides, while the anticodon is located at positions 34.35.36. Alanine in DNA is encoded by four codons: GCT, GCC, GCA, GCG. Therefore, 4 types of Ala tRNA are potentially possible, with anticodons AGC, GGC, TGC, CGC.
But the vast majority of the bacteria in question in the genome have only 2 species Ala tRNA_GCA and Ala tRNA_GCC. There are, of course, exceptions.
For analysis, we used sequenced DNA genomes that are available in the NCBI database. All identical non-mutated tRNAs were labeled with a unique identifier (Id). With the help of a computer program written for analysis, in a semi-automatic mode with manual verification, a list of various types of Ala tRNA was compiled , and their location in a particular sequenced locus.
Results
All examined strains of the genus Yersinia (9 pcs.) Have the same Ala tRNA_GCA with Id = 00046 and Ala tRNA_GCC with Id = 00043 in the genome. Based on this fact, we can conclude that indeed these strains have a strong evolutionary relationship, and therefore all of them belong to the genus Yersinia.
Now the genus Yersinia belongs to the Enterobacteriaceae family.But on the basis of the analysis done, within the framework of the Ala tRNA similarity, this relationship is unreasonable .
If you look at the classic representatives of the Enterobacteriaceae family, such as Escherichia, Salmonella, Shigella, Citrobacter, Cronobacter, Klebsiella, Pectobacterium, then they all have completely different Ala tRNAs. Namely, they have Ala tRNA_GCA with Id = 00011 and Ala tRNA_GCC with Id = 00012. On this basis, we can consider the listed genera of the Enterobacteriaceae family as classical representatives.
And only with the genus Photorhabdus, along the same line Ala tRNA_GCA with Id = 00046, Yersinia has a connection. Therefore, the genus Yersinia has traits from different families . We will call such a genus a transitional genus between different families .
So, if the Yersinia genus is associated with the Enterobacteriaceae family only by one configuration of the non-mutated Ala tRNA_GCA with Id = 00046 (and even partially), then the question arises of which family of the Yersinia genus is connected by another configuration of the non-mutated Ala tRNA_GCC with Id = 00043?
It turns out that the most direct relationship along this line is the genus Shewanella (family: Shewanellaceae, order: Alteromonadales, class: gamma-proteobacteria). At the same time, the Ala tRNA_GCC line connecting them with Id = 00043 is the key in evolutionary terms, since it is also present in the genera Pseudomonas and Vibrio. All these relations are closer than follows from the modern classification, where these genera are united only at the class level.
In turn, some representatives of the genus Shewanella have the same Ala tRNA_GCA with Id = 00047 and Ala tRNA_GCC with Id = 00043. Having already found out that Yersinia and Shewanella have a connection on one line (Id = 00043), it is interesting to whom the genus Shewanella is connected on another line (Id = 00047). It turns out that this line is also quite key in evolutionary terms. It branches further and Ala tRNA_GCA with Id = 00047 is also possessed by representatives of the genera Vibrio, Thiomicrospira, Saccharophagus.
We can also trace these connections further (see the figure). But now it’s clear that in order to systematize these evolutionary relations, it is necessary to slightly change the approach to their description.
higher resolution
{kind=link}
Conclusions
It should be emphasized once again that all the conclusions made in this work are based on the analysis of only tRNA Ala, and of course, for recognition of the results, verification of other types of tRNA is needed. But nevertheless, now we can draw some conclusions and describe how to change approaches to taxonomy.
Of sufficient complexity is the determination of which species is more evolutionarily ancient and which is younger. But based on the hypothesis that the biological world has evolved from simple to complex, we have at least one indisputable fact. If a bacterium has two chromosomes, then it seems obvious that it is evolutionarily more young than having one.
Therefore, as part of our study, we can confidently say that the genus Vibrio is younger than Shewanella or Yersinia. And then, since Vibrio and Shewanella are united identical by Ala tRNAGCA with Id = 00047, then with a high probability, the genus Vibrio descended from Shewanella and so one chromosome originated. After which other representatives of the genus Vibrio descended from the genus Colwellia, and so the second chromosome occurred. As a result of combining these chromosomes in one organism, we can talk about the genus Vibrio, which descended from Shewanella along one line and Colwellia along the other.
Thus, we should not speak of descent from one ancestor, but about at least two, or even more.
With monochromosomal bacteria, it is more difficult to determine the direction of evolution (who is younger and who is older). But based on two-chromosomal species, we can say, since there is a Vibrio species with Ala tRNA_GCA with Id = 00049 and Ala tRNA_GCC with Id = 00043; and there are also Vibrio species with Ala tRNA_GCA with Id = 00047, then Ala tRNA_GCA with Id = 00047 and Ala tRNA_GCC with Id = 00043 originally existed. And they were contained in Shewanella, and therefore it must be recognized as the most ancient organism, and put in the basis of the taxonomy of the organisms considered here.
Then we can conclude that from Shewanella in one line came Yersinia. From Yersinia, in turn, Photorhabdus, from which the entire family Enterobacteriaceae originated. But this is only one line. According to another, we already mentioned what kind of birth descended from Shewanella.
The multi-species origin greatly confuses the evolutionary picture, but there is nothing to be done about it - such is the complexity of speciation, and we only need to most accurately reflect them in conditions where not all species are known.
upd. Oh yes. I completely forgot, then they started talking - well, why should programmers fool their heads with all kinds of biology. So I actually wanted to interest programmers with this topic, since they are the ones who are able to write algorithms for bio-calculations. I just don’t have the strength to conduct a more complete analysis. Suddenly someone will be interested in mercy, please write in a personal.