Windows performance analysis using the capabilities of the OS and the PAL utility
Article author - Mikhail Komarov, MVP - Cloud and Datacenter Management
This article will cover:
- mechanism for working with performance counters;
- Configuring data collectors using both the graphical interface and the command line;
- creating a black box for recording data.
We also consider and discuss the work with the PAL utility and its application for data collection and analysis, including typical problems of localized systems.
In general, the performance problem can be represented in three parts: data collection, analysis of the data received and the creation of a black box for proactive monitoring of the problem system.
Data collection
Let's start with the long-known Performance Monitor. This is a standard utility that is included in all modern editions of Windows. Called either from a menu, or from the command line or search bar in Windows 8/10 by entering the perfmon command. After starting the utility, we see a standard panel in which we can add and remove counters, change the presentation and scale the graphs with data.
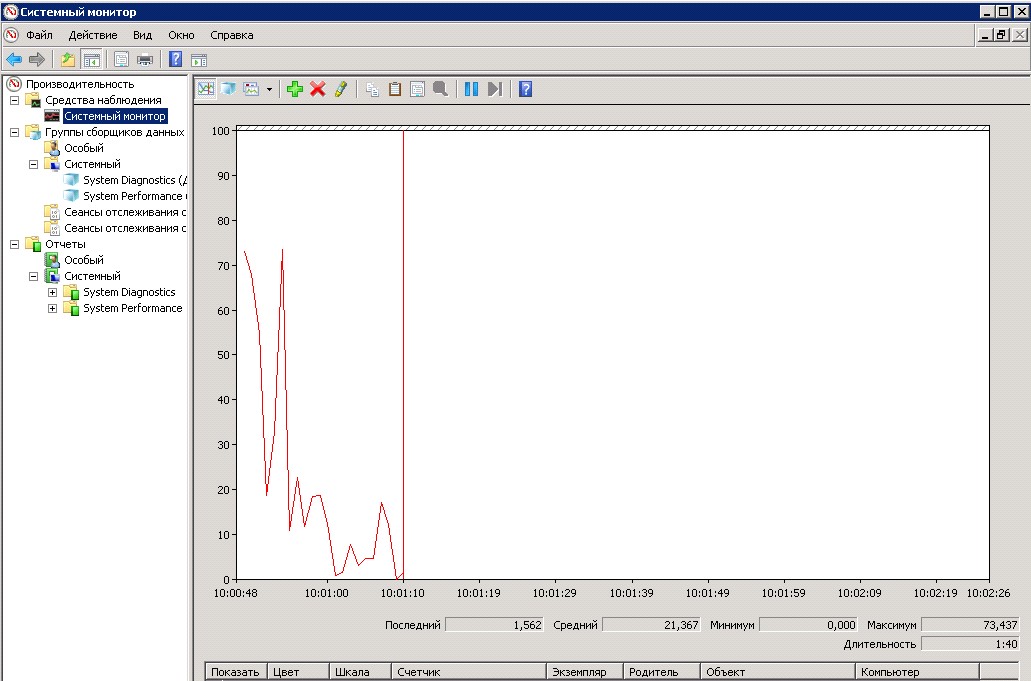
There are also data collectors, with the help of which data is collected on system performance. With certain skills and dexterity, operations to add counters and configure data collection parameters can be performed from the graphical interface. But when the problem arises of setting up data collection from several servers, it is more reasonable to use utilities with the command line. These are the utilities we will do.
The first utility is Typeperf , which can output data from performance counters to a screen or to a file, and also allows you to get a list of counters installed on the system. Examples of using.
Displays the processor load at 1-second intervals:
typeperf "\Processor(_Total)\% Processor Time"
Displays the names of performance counters associated with the PhysicalDisk object in a file:
typeperf -qx PhysicalDisk -o counters.txtIn our case, we can use the Typeperf utility to create a file with the counters we need, which we will use later as a template for importing counters into the data collector .
The next utility is Logman . This utility allows you to create, modify and manage various data collectors. We will create a data collector for performance counters. Here, for example, is a quick reference to the Logman command , which relates to performance counters and data collector management.
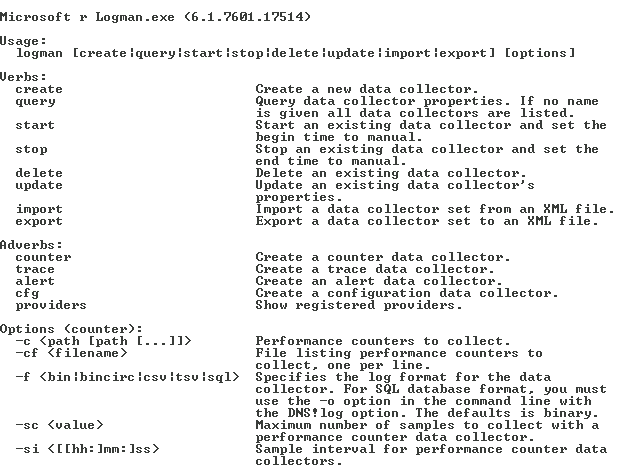

Let's look at a few examples that we will need in the future.
Create a data collector named DataCollector_test by importing performance counters from a filetest.xml :
logman import DataCollector_test -xml C:\PerfTest\test.xmlCreating a file for collecting performance data with circular mode enabled and a given size:
logman update DataCollector_test -f bincirc -max 600Changing the default performance data file path:
logman update DataCollector_test -o C:\PerfTest\Test_log.blgStarting the DataCollector_test data collector :
logman start DataCollector_testStopping the DataCollector_test data collector :
logman stop DataCollector_testNote that all these actions can be performed with a remote computer.
Consider another utility - Relog , which allows you to manipulate the data file after the data collector. Here is a description of it:

Below are several scenarios for using this utility.
Extracting performance counter data from the logfile.blg file using a filter with a list of counters counters.txt and writing the result in binary format:
relog logfile.blg -cf counters.txt -f binExtract the list of performance counters from logfile.blg to the text file counters.txt :
relog logfile.blg -q -o counters.txtWe will not work directly with this utility, but information about it will help in the future in case of problems in the PowerShell file generated by the PAL utility .
We note one more point, some analysis systems require data with the names of performance counters in English. If the interface of our system is in Russian, then we need to do the following manipulations: get a local user, give him permission to collect data (usually give local administrator rights), log in under it and change the interface language in the system properties.

Be sure to log out and log in a second time as this user to initialize the English interface and log out. Then indicate in the data collector that data will be collected on behalf of this user.

After that, the names of the counters and files will be in English.
Also note the ability to collect data for SQL Server using the utility from the product. This is SQLDIAG , which processes Windows performance logs, Windows event logs, SQL Server Profiler traces, SQL Server lock information, and SQL Server configuration information. You can specify what types of information should be collected using SQLdiag in the SQLDiag.xml configuration file .

You can use the PSSDIAG tool from codeplex.com to configure the SQLDiag.xml file . This is how the window of this tool looks. As a result, the data collection process for SQL may look like this. Using PSSDIAG, we create an xml file. Next, we send this file to the client, which launches SQLDIAG with our xml file on the remote server and sends us the result of the work as a blg file for analysis, which we will analyze in the next part.


Data Analysis with the PAL Utility
This utility is written by Clint Huffman, who is a Microsoft PFE engineer who analyzes system performance. He is also one of the authors of the authorized Vital Sign course, which is read at Microsoft and available to corporate customers, including in Russian in Russian. The utility is distributed freely, I will provide a link to it below.
This is what the utility start window looks like.

On the Counter Log tab, the path to the data file with the performance counters collected earlier is set. We can also set the interval for which the analysis will be performed.

On the Threshold File tabThere is a list of templates that can be exported in xml format and used as a list of counters for a data collector. Pay attention to the large selection of templates for performance analysis for various systems. An example of loading from the command line was shown above. The most valuable thing is that in these pre-prepared templates, boundary values are set for these parameters, which will be used in the future to analyze the collected data !!!
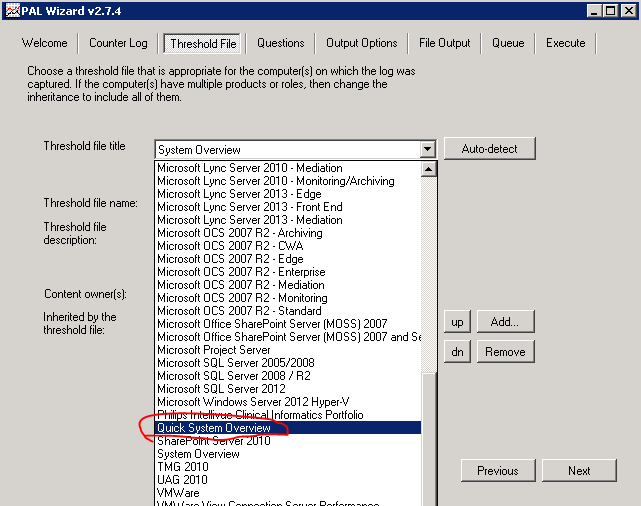

Here, for example, the boundary values for disk performance counters look like:

We can create our own templates using the necessary counters, which will be tailored to the needs of our organization.
We proceed according to the following algorithm: on the workstation, run the PAL utility , go to the tabThreshold File and export the template as an xml file. Based on this file on the server, we create a data collector and start the collection of information.
After collecting the data, copy the resulting file to the workstation so that the analysis does not load the server, return to the Counter Log tab , specify the path to the file. Again, go to the Threshold File and select the same template that was exported for the data collector.
We switch to the Question tab and indicate the amount of RAM on the server on which the data was collected. In the case of a 32-bit system, fill UserVa .

Go to the Output Options tabon which we set the partition interval for analysis. The default value AUTO divides the interval into 30 equal parts.

The File Output tab looks quite usual, we indicate on it the path to the final report files in HTML or XML format.

The Queue tab shows the resulting PowerShell script. In general, we can say that the utility collects the parameters that it substitutes into the PAL.PS1 script .

The summary tab sets the execution parameters. You can run multiple scripts at once and specify the number of threads on the processor. I would like to emphasize that blg processing is not done by the utility, but by the PowerShell script, and this opens up possibilities for fully automated log analysis. For example, the data collector is restarted every day, as a result, the current blg-file is freed and a new one is created. The old file is copied to a special server, where the script processing this file will be launched. After that, the finished HTML or XML file with the results is moved to a specific directory or sent to the mailbox.

Please note that the utility should work only in English localization. Otherwise, we get an error message.

Also, the data file should be with the names of the meters in English. I have indicated how to do this above. After clicking Finish, a PowerShell script will be launched, the operating time of which depends on the amount of data and the speed of the workstation.
The result of the utility will be a report in the selected format, in which there are graphs and numerical data that allow you to understand what happened in the system for a given period, taking into account the boundary values of alerts in the template on the Threshold File tab. In general, the analysis of the HTML file will allow at the initial stage to identify problem areas in the system and understand where to go next, both in terms of finer monitoring, and in terms of upgrading or reconfiguring the system. The blog of Clint Huffman has a script that can convert a template file with boundary conditions into a more understandable format.


Black box
Sometimes there is a need for proactive monitoring of a problem system. To do this, we will create a “black box” in which we will write performance data. Back to the scripts described earlier.
Create a data collector named BlackBox by importing performance counters from the SystemOverview.xml file , which you unloaded from the PAL utility or created yourself:
logman import BlackBox -xml C:\ BlackBox\SystemOverview.xmlCreating a file for collecting performance data with circular mode enabled and a specified size of 600 MB (about 2 days with a standard set of counters):
logman update BlackBox -f bincirc -max 600Changing the default performance data file path:
logman update BlackBox -o C:\ BlackBox \ BlackBox _log.blgStarting the BlackBox Data Collector:
logman start BlackBoxThis script creates the task of restarting the data collector in the event of a system restart:
schtasks /create /tn pal /sc onstart /tr "logman start BlackBox " /ru systemJust in case, we’ll fix the properties of the data manager so as not to fill the disk space, since after restarting the data collector a new file with a limit of 600 MB is created.

Note that you can copy the data file only when the data collector is stopped. You can stop the latter with a script or using the graphical interface.
Stopping the BlackBox Data Collector:
logman stop BlackBoxThis concludes the part on collecting and initial performance analysis.
Resources
Performance Monitor
https://technet.microsoft.com/en-us/library/cc749154.aspx
PAL Utility
https://pal.codeplex.com/
Clint Huffman Blog
http://blogs.technet.com/b/clinth/
Clint Huffman
Windows Performance Analysis Field Guide
http://www.amazon.com/dp/0124167012/ref=wl...=I2TOVTYHI6HDHC