Systematics of prokaryotes - distant relatives
In the summer, I planned an experiment and wrote an article Using UML for an experiment on the evolutionary systematics of prokaryotes, and indirectly about the psychology of scientists . The rough processing results were ready by the end of the summer (thanks, mktums for the help).
Now a pause has formed, and I finished off this topic, and present the results.
(I’ll repeat something from the previous article so as not to force new readers to read it)
The main criticism of the article Interesting results about the evolutionary systematics of prokaryotes or “multi-species origin” consisted in the following claim “ You cannot consider one gene as a measure ”. I completely agree with this, and this experiment completely corrects this.
A few numbers. Now in the NCBI there are about 2,000 bacterial genomes (3,723 loci). In preparation for the experiment, I selected all the tRNAs that are labeled in this way. They turned out to be more than 40 thousand unique variations. But alas, there are a lot of mistakes among them (about 50%, see previous articles, where this was discussed in detail).
But I thought that you can skip the stage of full error correction. How to do it? I sorted these tRNAs by length and by the presence of the end of CCA at the end of the sequence. I must say that the CCA sequence is required for any tRNA, and the length can be from 74 to 96 nucleotides.
There are many miracles in the NCBI right down to tRNA from a single nucleotide, or more than 1300 :) (you can't say without a smile). Therefore, I removed sequences that have a length of up to 70 and greater than 100, as well as those that do not end in CCA.
There are about 20,000 of them. These are the most probable tRNAs that do not contain errors from the NCBI. With the remaining half of tRNA - you can figure it out later.
In fact, for the planned experiment, it makes no difference whether this particular sequence contains 70-100 nucleotides in length or not. Why? Since I am going to double-check the genomes of 2000 bacteria, are there really such sequences - errors will be excluded. And tRNA is actually the second thing or not. The main thing is that significant organisms of DNA coincide in different organisms. The coincidence of the sequence length of 70-100 in the genomes is far from accidental.
Therefore, what am I doing now. I take these 20,000 tRNAs and find in which bacteria they are present. If the sequence is present in only one organism, this is not interesting. And most likely this is an erroneous sequence. And thus a substantial percentage of errors are eliminated.
If the sequence is in more than one organism, this is one association (connection) between two organisms.
The first article made an important conclusion that
I was also advised to display the graph using Graphviz, which I did. But Graphviz freezes when the number of links in the graph is more than 1000. And I got a total graph for 6172 links. Therefore, here I show only a small fragment for clarity. And I give a link to a graph of almost 1000 links.
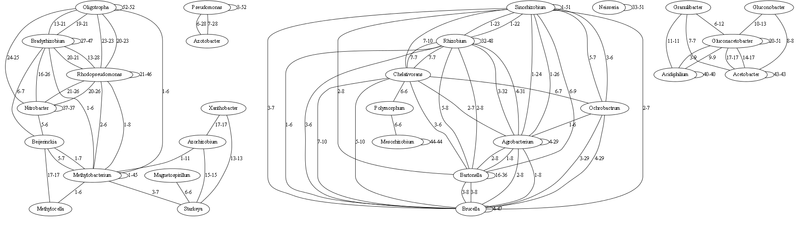
Here is the graph with the strongest bonds (links to 5 identical tRNAs inclusively omitted)
Each link is characterized by the minimum-maximum number of matching (100% identical) tRNA genes. The relationship of a genus with itself means the number of identical tRNAs within this genus (i.e., how species differ).
In fact, all this still needs to be visually processed so that it is possible to visually embrace all this multitude. On a graph with 1000 links, there are many genera that are not connected with anyone - but if we were to display weaker links with up to 5 identical tRNAs, then we could see distant relatives. (I’m thinking of doing it the next step, if there are people who want to help - write).
In fact, on this basis, much coincides according to the current classification. The number of identical tRNAs well illustrates the range of genera from each other, the less identical tRNAs - the more the ancient ancestor. Those genera that have few connections are the most ancient (because they are sequencing now, and their population is currently represented by separate species). By analyzing them, one can build quite accurately the process of initial evolution.
upd.Removed two-way connections from the graph (clogged the image). The total number of links decreased to 4551. This allowed us to display a larger graph:
You can download the image here (11.2 MB). Here is the graph with the strongest bonds (links to 3 identical tRNAs are omitted inclusively).
Then the bonds (intermediate species) between two huge domains (visible on the image, presumably correspond to Beta and Gamma proteobacteria), and other details are visible. How much does this correspond to the current classification should be compared, but there is something to think about (just the detail is such that there is probably something that did not fall into the current scientific classification).
upd2 Using yEd Graph Editor it turned out to display the full graph. Below is a mini picture.

The image turns out badly because of the connections, details are not visible, therefore the file is in the yEd Graph Editor format below, at least you can enlarge, move and make out. If someone is interested and makes a more visible graph - I will say thanks :).
Count “Systematics of prokaryotes (505 genera and 4548 connections between them)”
Now a pause has formed, and I finished off this topic, and present the results.
Method
(I’ll repeat something from the previous article so as not to force new readers to read it)
The main criticism of the article Interesting results about the evolutionary systematics of prokaryotes or “multi-species origin” consisted in the following claim “ You cannot consider one gene as a measure ”. I completely agree with this, and this experiment completely corrects this.
A few numbers. Now in the NCBI there are about 2,000 bacterial genomes (3,723 loci). In preparation for the experiment, I selected all the tRNAs that are labeled in this way. They turned out to be more than 40 thousand unique variations. But alas, there are a lot of mistakes among them (about 50%, see previous articles, where this was discussed in detail).
But I thought that you can skip the stage of full error correction. How to do it? I sorted these tRNAs by length and by the presence of the end of CCA at the end of the sequence. I must say that the CCA sequence is required for any tRNA, and the length can be from 74 to 96 nucleotides.
There are many miracles in the NCBI right down to tRNA from a single nucleotide, or more than 1300 :) (you can't say without a smile). Therefore, I removed sequences that have a length of up to 70 and greater than 100, as well as those that do not end in CCA.
There are about 20,000 of them. These are the most probable tRNAs that do not contain errors from the NCBI. With the remaining half of tRNA - you can figure it out later.
In fact, for the planned experiment, it makes no difference whether this particular sequence contains 70-100 nucleotides in length or not. Why? Since I am going to double-check the genomes of 2000 bacteria, are there really such sequences - errors will be excluded. And tRNA is actually the second thing or not. The main thing is that significant organisms of DNA coincide in different organisms. The coincidence of the sequence length of 70-100 in the genomes is far from accidental.
Therefore, what am I doing now. I take these 20,000 tRNAs and find in which bacteria they are present. If the sequence is present in only one organism, this is not interesting. And most likely this is an erroneous sequence. And thus a substantial percentage of errors are eliminated.
If the sequence is in more than one organism, this is one association (connection) between two organisms.
results
The first article made an important conclusion that
The multi-species origin greatly confuses the evolutionary picture, but there is nothing to be done about it - such is the complexity of speciation, and we only need to reflect them most accurately in conditions when not all species are known.
And therefore, for an adequate description, trees are not phylogenetically needed. At a minimum, we can talk about genital trees with two parents (for averaging), and in the general case, a graph.
I was also advised to display the graph using Graphviz, which I did. But Graphviz freezes when the number of links in the graph is more than 1000. And I got a total graph for 6172 links. Therefore, here I show only a small fragment for clarity. And I give a link to a graph of almost 1000 links.
Here is the graph with the strongest bonds (links to 5 identical tRNAs inclusively omitted)
{kind=link}
Each link is characterized by the minimum-maximum number of matching (100% identical) tRNA genes. The relationship of a genus with itself means the number of identical tRNAs within this genus (i.e., how species differ).
Some conclusions
In fact, all this still needs to be visually processed so that it is possible to visually embrace all this multitude. On a graph with 1000 links, there are many genera that are not connected with anyone - but if we were to display weaker links with up to 5 identical tRNAs, then we could see distant relatives. (I’m thinking of doing it the next step, if there are people who want to help - write).
In fact, on this basis, much coincides according to the current classification. The number of identical tRNAs well illustrates the range of genera from each other, the less identical tRNAs - the more the ancient ancestor. Those genera that have few connections are the most ancient (because they are sequencing now, and their population is currently represented by separate species). By analyzing them, one can build quite accurately the process of initial evolution.
upd.Removed two-way connections from the graph (clogged the image). The total number of links decreased to 4551. This allowed us to display a larger graph:
You can download the image here (11.2 MB). Here is the graph with the strongest bonds (links to 3 identical tRNAs are omitted inclusively).
{kind=link}
Then the bonds (intermediate species) between two huge domains (visible on the image, presumably correspond to Beta and Gamma proteobacteria), and other details are visible. How much does this correspond to the current classification should be compared, but there is something to think about (just the detail is such that there is probably something that did not fall into the current scientific classification).
upd2 Using yEd Graph Editor it turned out to display the full graph. Below is a mini picture.
The image turns out badly because of the connections, details are not visible, therefore the file is in the yEd Graph Editor format below, at least you can enlarge, move and make out. If someone is interested and makes a more visible graph - I will say thanks :).
Count “Systematics of prokaryotes (505 genera and 4548 connections between them)”